层状可调铁掺杂氮化碳及其光芬顿催化性能研究
2022-05-23李琛,周烈兴,李雪梅,解林坤,柴希娟*
李 琛, 周 烈 兴, 李 雪 梅, 解 林 坤, 柴 希 娟*
(1.西南林业大学 国家生物质材料国际联合研究中心,云南 昆明 650224;2.昆明理工大学 分析测试研究中心,云南 昆明 650093;3.西南林业大学 化学工程学院,云南 昆明 650224 )
0 引 言
在全球环境污染问题日益严重、能源短缺日益凸显的今天,光催化技术因安全高效、绿色环保、无二次污染等优点而在降解有机污染物、光解水制氢制氧、有机合成等方面备受关注.在以往研究的众多光催化材料中,以三均三嗪为基本结构单元的类石墨相氮化碳(g-C3N4),因其理化性质稳定、禁带宽度较窄(2.7 eV)、对可见光响应、无毒、电子结构独特且易于调控等优点,成为近些年来光催化领域的研究热点[1-3].然而,由常规热缩聚法制备的g-C3N4多呈致密的块状堆积结构,其比表面积小、光生载流子寿命短、电子-空穴对易快速复合等缺点极大地限制了氮化碳在光催化领域的应用.相比之下,g-C3N4二维多孔纳米片结构展示出较高的比表面积和更多的活性位点.同时,二维纳米片的超薄结构还极大地缩短了光生电子和空穴迁移到材料表面的路径.目前,已有一些成功制备g-C3N4二维纳米片的方法,如液相剥离法[4]、机械剥离法[5]、超临界剥离法[6]和热剥离法[7-9]等.在这些方法中,热剥离法被认为是制备高质量g-C3N4薄层的最有效和快速的方法.在加热过程中,g-C3N4的含氧官能团分解并产生大量气体,产生足够的压力以克服石墨烯片之间的范德华力,并进一步扩展片层以形成多孔框架[10].
多种改性方法如元素掺杂法(Fe、P、Cu和B)[11-14]、贵金属表面修饰(Ag、Au)[15-16]、与其他半导体复合形成异质结(TiO2、WO3)[17-18]等亦常用于提高g-C3N4光催化反应效率.其中,元素掺杂法是改善g-C3N4电子结构和表面性质的较简单有效的手段.到目前为止,铁元素作为最有前景的掺杂元素之一,在g-C3N4掺杂方面受到越来越多的关注[19].Fe3+还被广泛应用于芬顿(Fenton)反应中.芬顿反应是一种高效且经济的废水高级氧化技术,在酸性条件下,均相溶液体系中的Fe2+可以催化分解双氧水,产生羟基自由基[20].羟基自由基具有强氧化性和高反应活性,可以无选择性地氧化降解有机污染物.然而,液相铁离子导致的铁泥、腐蚀性反应环境(一般pH<3)及无法实现活性组分与反应溶液分离等问题限制了均相芬顿体系在污水处理方面的广泛应用[21].较均相芬顿体系而言,多相芬顿催化剂因具有pH工作范围温和、不产生铁泥等优点而受到研究者的关注[22],其最大的特点是固相化自由金属离子,形成固体催化剂.
本文以二氰二胺为前驱体,九水硝酸铁为铁源,通过调节煅烧时间和掺杂比例制备一系列热剥离型铁掺杂g-C3N4(Fe-CN),考察铁掺杂量和热剥离时间对Fe-CN结构、形貌的影响.同时以亚甲基蓝(MB)溶液为模拟污染物,通过外加H2O2构建光催化芬顿/光芬顿协同体系,考察中性环境下固相Fe-CN在芬顿体系中催化性能,筛选性能高效的Fe-CN光催化材料.
1 实 验
1.1 试剂与仪器
二氰二胺、九水硝酸铁、无水乙醇、过氧化氢(97%)、亚甲基蓝均为分析纯,购自国药集团化学试剂有限公司.
仪器包括X射线衍射仪(XRD,LabX6000型)、扫描电镜(SEM,Apreo型)、透射电镜(TEM,Tecnai-10型)、物理吸附仪(BET,BETA201A 型)、紫外-可见分光光度计(Lambda750)、傅里叶红外光谱仪(FT-IR,ALPHA 型)、X射线光电子能谱仪(XPS,AXIS ULTRA DLD型).
1.2 样品制备
1.2.1 g-C3N4的制备 以二氰二胺为前驱体,将装有10 g前驱体的石英舟置于管式炉中部,在氮气气氛中550 ℃恒温煅烧4 h,升温速率为10 ℃/min.反应结束后,样品自然冷却至室温,充分研磨后得到淡黄色g-C3N4,记作CN.
1.2.2 Fe-CN的制备 分别称取0.014 4、0.043 4、0.072 3、0.101 3 g Fe(NO3)3·9H2O溶于40 mL去离子水中,加入2 g CN,超声15 min 后置于100 ℃油锅中,使水分缓慢蒸干.将得到的固体放入100 ℃烘箱中干燥6 h,研磨后放入石英舟中,再置于管式炉中于550 ℃条件下焙烧2~4 h.气氛为空气,升温速率为5 ℃/min.反应结束后自然冷却后取出,得到不同掺杂比例和不同焙烧时间的Fe-CN催化剂,将所得产物研磨至细腻均匀,记作xFe-y,其中x表示铁离子的质量分数,y表示焙烧时间.
1.3 光催化性能测试
选取10 mg/L的亚甲基蓝溶液作为模拟污染物.准确称量0.02 g样品于100 mL模拟污染物中,在无光环境下充分搅拌30 min,使吸附-脱附达到平衡.在体系中加入0.2 mL H2O2开启芬顿反应,每隔10 min取样3 mL,放入离心机中以10 000 r/min离心5 min,取上层清液测其吸光度.相同实验条件下,使用350 W氙灯作为光催化反应的辐照光源进行光芬顿反应.
2 结果与讨论
2.1 样品表征
为了表征样品的形貌,分别对其进行扫描电镜和透射电镜分析,如图1所示.其中,图1(a)和(c)分别为由直接热缩聚法制备得到CN的SEM和TEM照片.从图1(a)可以看出,由直接热缩聚法得到的CN为片状堆叠的块状颗粒,片层之间连接紧密,呈现明显的团聚现象.由图1(c)可以看出,CN呈现典型的片状结构,颜色深浅不一是由于片状堆积的层数不同引起的,且片层上无孔洞结构.图1(b)和(d)分别为铁掺杂量为0.5%、热剥离时间为3 h时样品0.5%Fe-3h的SEM和TEM照片.由图1(b)可以看出,样品0.5%Fe-3h呈现明显的木耳状片层结构,片层厚度薄且片层间无明显的堆积现象,整体结构蓬松.图1(d)显示0.5%Fe-3h呈片层结构,且片层较薄,片层上可以观察到少量的孔洞结构.0.5%Fe-3h的SEM和TEM照片均表明,铁离子的引入使得CN粉体的微观形貌发生了变化,同时样品表面多孔结构增加.这可能是由于掺杂铁后在二次高温焙烧过程中铁对片层结构的腐蚀及CN的含氧官能团分解产生的气体对片层的冲击造成的.薄层结构和多孔结构有利于增大CN的比表面积,增加反应活性位点,使污染物可以更好地吸附在材料表面,从而更有利于污染物的降解,提高其光催化性能[10].
CN和Fe-CN的XRD谱图如图2所示.由图可知,纯CN及不同条件下制备得到的Fe-CN均在13.9°和27.5°处出现两个特征衍射峰,对应CN的(100)和(002)晶面,分别是由石墨相层间堆垛的周期性排列和层内共轭芳香物层间堆积引起[23].掺铁后的Fe-CN与CN的特征衍射峰大致相同,说明铁掺杂并没有改变CN的基本结构单元.从图2可以看出,随着铁掺杂量的增加,Fe-CN在(002)晶面处吸收峰由尖锐逐步趋于平缓,同时峰位向高角度移动.这是由于铁掺杂减弱了石墨相氮化碳层间的范德华力,使层间距变宽,晶粒尺寸减小,结晶度降低.同样,Fe-CN在(100)晶面处的峰强也明显减弱,说明氮化碳面内的三均三嗪环周期性排列的规整性降低,这与掺铁后片层上形成的孔洞结构有关.对比不同二次焙烧时间下Fe-CN的XRD谱图可以发现,随着焙烧时间的延长,Fe-CN在(100)和(002)晶面处的峰强均有所减弱.图2中未发现单质铁、氧化铁、碳化铁等铁物种衍射峰,这表明铁可能以金属卟啉或者金属酞菁中Fe—N键的形式掺杂到CN的骨架中[24-25].

(a)CN的SEM

图2 CN和Fe-CN的XRD谱图Fig.2 XRD patterns of CN and Fe-CN
二次煅烧时间为3 h的Fe-CN和CN的FT-IR 谱图如图3所示.在CN的红外光谱中,吸收峰主要集中在808、1 240~1 650和2 800~3 400 cm-13个区域,分别对应三嗪环结构的弯曲振动峰[26]、芳香碳氮杂环化合物的伸缩振动峰[27]和N—H键的伸缩振动峰[28].xFe-3h样品的红外特征吸收峰与CN基本一致,没有观察到新的特征峰,说明铁掺杂并没有改变CN的基本骨架结构.这是由于掺杂进入CN体相的铁离子与CN层内的吡啶氮原子形成配位键,铁被锚定在七嗪环内,故而不影响CN的基本骨架结构[24].
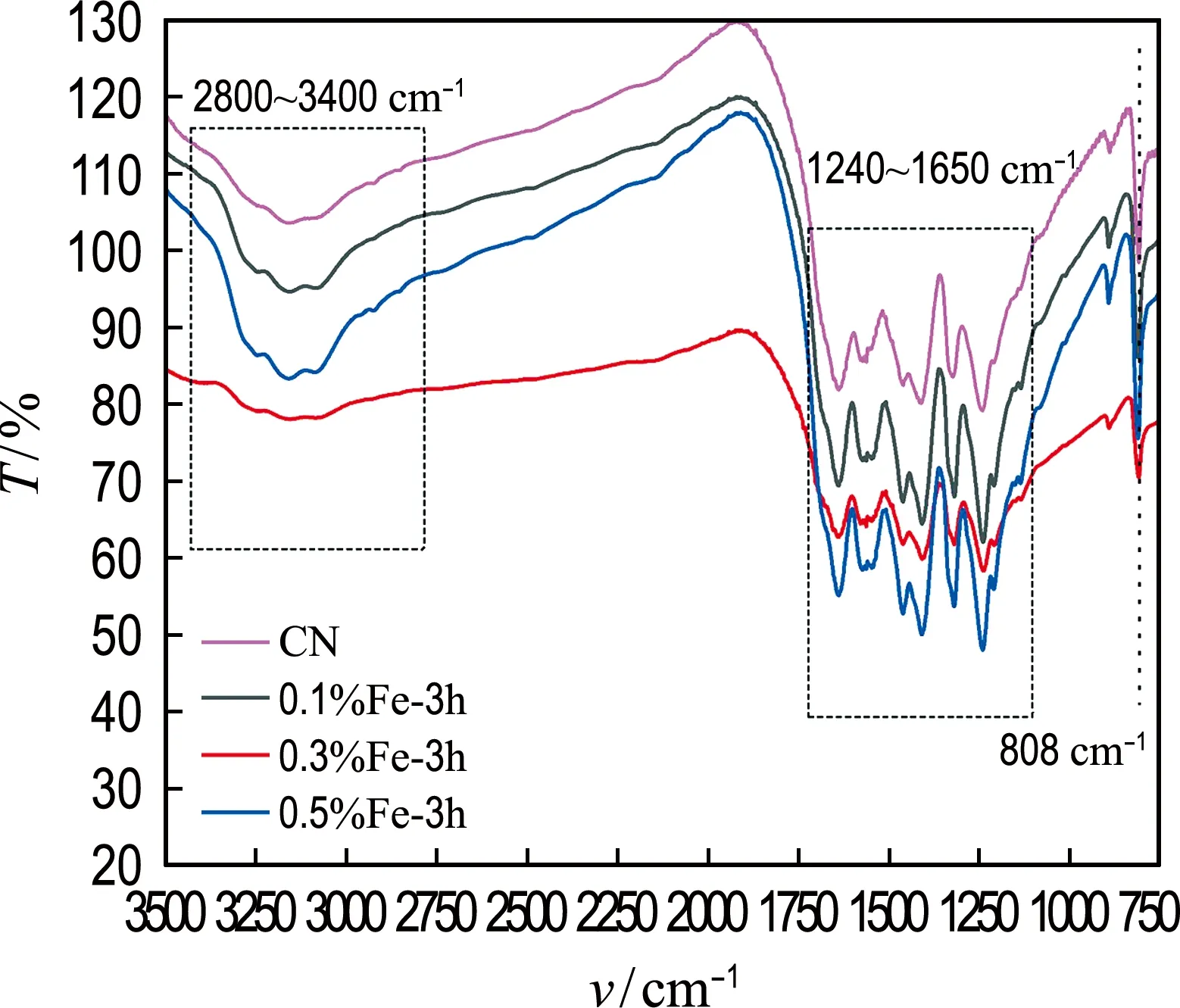
图3 xFe-3h和CN的FT-IR谱图Fig.3 FT-IR spectra of xFe-3h and CN
表1为CN和Fe-CN的BET测试结果.从表1可以看出,掺杂铁后CN的比表面积明显增大,且当铁掺杂量为0.5%时,0.5%Fe-3h的比表面积达最大值74.024 1 m2/g,是CN的3.89倍.这是因为,铁的掺杂使氮化碳呈现出明显的木耳状片层结构,片层变薄,CN颗粒粒径减小,团聚现象明显弱化,从而使得样品的比表面积增加.但当铁掺杂量过多时,Fe3+难以进入CN晶格中,因此反而使得样品的比表面积有所减小[29-30].从图4(a)可以看出,CN和0.5%Fe-3h样品对应的等温吸附曲线均为Ⅲ型[31],但在低压端(p/p0=0~0.1)CN较0.5%Fe-3h 更靠近横坐标,且相对平缓,说明CN与氮气间的相互作用力较0.5%Fe-3h弱.这是因为0.5%Fe-3h样品的片层结构上有孔洞存在,N2分子以单层或多层吸附于孔内,使得其在低压时依靠微孔自身的强吸附能力而存在一定的吸附量.由表1可知,Fe-CN的孔径为9~14 nm.由图4(a)可以看到0.5%Fe-3h呈现出明显而细长的H3型滞后环,而H3型的孔隙多为层状结构引起的狭缝孔,对应于石墨相氮化碳的片层结构.对比不同焙烧时间下样品的比表面积可以发现,焙烧时间的增长不会持续增加样品的比表面积.这是因为随着焙烧时间的延长,氮化碳片层上的孔洞尺寸持续增大,部分片层结构发生崩解[32].

表1 CN和Fe-CN的比表面积、孔容、孔径Tab.1 Specific surface area,pore volume and pore diameter of CN and Fe-CN

(a)吸附-脱附曲线
图5为所制备样品的UV-Vis漫反射谱图及hν-(ahν)1/2图.由图5(a)可知,掺杂铁后,Fe-CN的吸收边发生显著红移,同时其在300~700 nm的光吸收强度也显著提高.说明铁的掺入拓宽了CN的光吸收范围和吸收强度,提高了光利用率.由半导体禁带宽度求导公式求得CN和0.5%Fe-3h禁带宽度分别为2.72 eV和2.27 eV,说明铁的引入改变了CN的能带结构,降低了其带隙能.带隙的减小进一步说明了Fe-CN具有比CN更宽的光响应范围和更高的光利用率,因此表现出优异的光催化活性.


(a)UV-Vis漫反射谱图
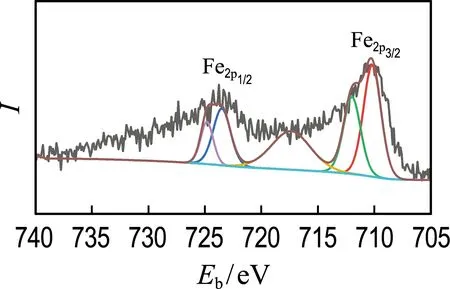
图7是样品0.5%Fe-3h在光芬顿和暗芬顿循环3次后的Fe2pXPS谱图.根据XPS可以确定各样品中Fe2+与Fe3+的比例.在新鲜材料、3次光芬顿和3次暗芬顿后的样品中,Fe2+与Fe3+的比例分别为1.57、1.52和0.98.这是因为在光芬顿反应中,Fe3+能够捕获CN中的光生电子被还原为Fe2+,同时Fe2+可被H2O2氧化为Fe3+,从而保持体系中Fe2+/Fe3+的动态平衡.而在暗芬顿反应中,Fe2+被H2O2氧化为Fe3+后,Fe3+不能捕获CN中的光生电子,无法被还原为Fe2+.因此,在经历暗芬顿反应后Fe2+与Fe3+的比例下降.

(a)C1s
(a)光芬顿
2.2 光催化性能
图8(a)是CN和xFe-3h样品随光照时间变化降解MB溶液的活性曲线,其中0.5%Fe-3h的光催化性能最佳(降解率为72%,是CN降解率的1.67倍).0.5%Fe-3h较薄的片层和孔洞结构使得光生电子和空穴迁移到颗粒表面的距离大大缩短,有利于提高光生载流子的分离效率.同时,较薄的片层和孔洞结构能促进入射光在层间的多次反射,进而明显提高材料对光的吸收,从而产生更多的光生电子-空穴对[35].当铁掺杂量大于0.5%时降解率反而出现下降,这是由于铁掺杂量过多使得Fe3+难以进入CN晶格[36].图8(b)为各样品降解MB溶液的一级反应速率常数k,其中样品0.5%Fe-3h的k最大,为0.011 1 min-1,是纯CN的2.41倍.
图8(c)、(e)是0.5%Fe-y样品进行光芬顿与暗芬顿降解MB溶液的活性曲线,其对应的一级反应速率常数分别见图8(d)、(f).光芬顿下0.5%Fe-3h 样品的降解率为99.8%,是纯CN的2.32倍,0.5%Fe-3h的铁离子对H2O2的分解具有类似芬顿的催化活性,可见光照下的降解率远高于无光照下的降解率(83%,是纯CN的1.93倍).0.5%Fe-3h光芬顿在所有样品中具有最高的活性,这表明铁掺杂可以增强类光芬顿活性.同时电子在Fe和CN轨道上的转移降低了电子-空穴对的复合,并且均匀分布在CN的Fe3+为H2O2的吸附提供了较高的比表面积.对于暗条件下的样品,不存在半导体光催化过程中的Fe与CN间的电子转移,Fe2+仅被H2O2氧化成Fe3+,使得Fe2+与Fe3+比例下降,反应活性与稳定性下降.而在光芬顿反应中,Fe3+迅速捕获CN光催化反应生成的光生电子被还原为Fe2+,使得Fe2+与Fe3+比例趋于稳定,提升了催化剂的反应活性与稳定性[36].
2.3 催化机理分析
根据以上分析测试结果,提出了可见光促进Fe-CN光芬顿反应性能的机理.图9为0.5%Fe-3h光芬顿降解MB溶液反应机理图.在光芬顿过程中,与H2O2反应形成芬顿体系的Fe2+来源有两种.一是在可见光照射下热剥离型Fe-CN被激发产生光生电子(e-)和光生空穴(h+)(式(1)).光生电子被锚定在氮化碳七嗪环骨架上的Fe3+捕获并还原为Fe2+(式(2))[36].二是Fe3+与体系中的H2O2反应生成Fe2+和超氧酸自由基(HO2•)(式(3))[30].以上两种路径为体系中的Fe2+提供了丰富的来源.Fe2+通过与系统中外加的H2O2进一步反应生成Fe3+和具有强氧化性的羟基自由基(•OH)(式(4)),氧化降解MB(式(7))[37].以上Fe2+/Fe3+的动态循环保证了光芬顿体系中Fe2+/Fe3+的氧化还原动态平衡,提高了光芬顿反应速率.因此,Fe2+的快速再生以及保持体系稳定的Fe2+与Fe3+比例是Fe-CN催化剂在光芬顿反应中具有较高活性和稳定性的原因.与此同时,式(3)中生成的HO2•发生相互反应生成O2和H2O2继续参与反应(式(6))[38].
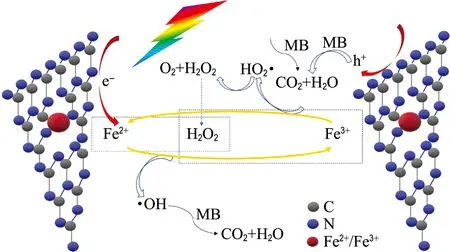
图9 0.5%Fe-3h 光芬顿降解MB溶液反应机理图Fig.9 Photo-Fenton reaction mechanism diagram of 0.5%Fe-3h degrading MB solution


(a)光催化反应性能
Fe-CN+hν→e-+h+
(1)
Fe3++e-→Fe2+
(2)
Fe3++H2O2→Fe2++HO2·+H+
(3)
Fe2++H2O2→Fe3++·OH+OH-
(4)
(5)
2HO2·→H2O2+O2
(6)
(7)
3 结 论
(1)以二氰二胺为前驱体,九水硝酸铁为铁源,通过两步法制备了一系列不同铁掺杂比例的热剥离型Fe-CN.光催化结果表明,铁掺杂量为0.5%,热剥离时间为3 h时的样品0.5%Fe-3h的光催化效果最优,其在光催化体系、暗芬顿体系和光芬顿体系中,60 min时光催化降解亚甲基蓝(MB)溶液的降解率分别为72%、83%和99.8%,为纯CN的1.67、1.93和2.32倍.
(2)对Fe-CN的结构分析表明,铁离子被成功引入氮化碳的结构中,氮化碳从致密的块状堆积结构转变为独特的木耳状片层介孔结构,片层结构明显变薄,且呈现出一定的多孔结构,样品0.5%Fe-3h 的比表面积是CN的3.89倍.同时,铁掺杂并没有改变CN的骨架结构,且在二次焙烧过程中,掺杂的Fe3+被还原为Fe2+.
(3)在光芬顿过程中,Fe2+来源有两种.一是掺杂的Fe3+捕获光生电子后被还原为Fe2+,二是Fe3+与H2O2反应生成Fe2+.同时,Fe2+通过与H2O2反应生成Fe3+.从而保证了光芬顿体系中Fe2+/Fe3+的氧化还原动态平衡,提高了光芬顿反应速率.
