线粒体脑肌病的MRI诊断
——2022年读片窗(5)
2022-05-21王龙胜
王龙胜
作者单位: 230601 安徽合肥 安徽医科大学第二附属医院放射科
1 病史摘要
患者,女性,55岁,1周前患者无明显诱因下出现胡言乱语、不认识家人、理解不能,无四肢无力、头晕头痛、饮水呛咳,症状进行性加重。病程中患者饮食睡眠可,大小便正常,近期体质量未见明显下降。既往有糖尿病病史。体格检查:体温36.0℃ 、脉搏83次/分、 呼吸20次/分、 血压 124/92 mmHg (1 mmHg≈0.133 kPa),神清,精神一般,反应迟钝,自言自语,问话不答,双侧瞳孔等大同圆,四肢肌力检查不配合,能自行行走。病理征不配合,病程中患者饮食睡眠可,大小便正常,近期体质量未见明显下降。实验室检查:C-反应蛋白<0.5 mg/L,白细胞计数6.71×109/L,血红蛋白142 g/L,血小板计数274×109/L,血浆乳酸2.9 mmol/L↑。脑脊液生化:葡萄糖7.26 mmol/L↑,氯122.8 mmol/L,腺苷脱氨酶1 U/L,脑脊液总蛋白483 mg/L↑。 脑脊液常规:潘氏试验弱阳性↑,白细胞35×106/L↑。脑脊液墨汁染色未检出新型隐球菌。脑脊液乳酸:乳酸3.8 mmol/L↑。
2 MRI检查所见
双侧枕叶可见对称斑片状长T1、长T2信号(图1、2),FLAIR序列呈高信号(图3),DWI序列呈高信号(图4),ADC图呈稍低信号(图5),测ADC值约0.76×10-3mm2/s;脑室系统扩大,脑沟裂明显增宽,中线结构居中,幕下小脑及脑干未见明显异常信号。MRS右侧枕叶病灶显示NAA峰及CHO峰稍下降,可见明显峰倒置的Lac(图6)。

图1 T1WI

图2 T2WI

图3 FLAIR

图4 DWI

图5 ADC
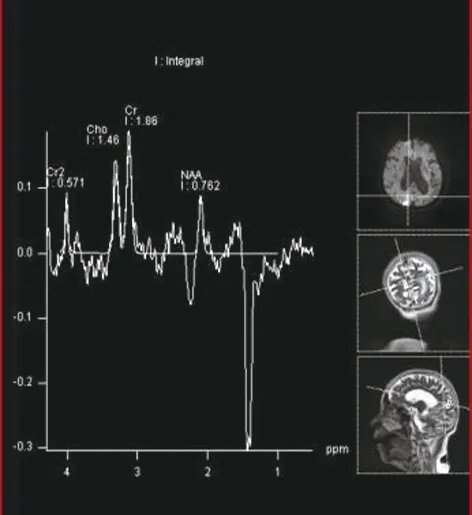
图6 MRS
3 临床诊断
线粒体DNA高敏感测序分析结果阳性,临床诊断为线粒体脑肌病。
4 讨论
线粒体脑肌病是由于线粒体遗传物质DNA有缺陷引起的一组与线粒体氧化磷酸化功能异常有关的遗传代谢性疾病,有多种类型,包括肌阵挛性癫伴破碎红纤维综合征(myoclonus epilep- sy with ragged-red fibers,MERRF)、线粒体脑肌病伴高乳酸血症及卒中样发作、慢性进行性眼外肌瘫痪综合征(chronic progressive external othalmoplegia,CPEO)、Leigh综合征(leigh syndrome,LS)、Kearne-Sayre综合征(Kearne-Sayre syndrome,KSS)等,其中线粒体脑病是最常见的一种。它是一种慢性进行性的神经退行性疾病,脑部病理改变为脑组织海绵样变性、灶状坏死、脱髓鞘伴铁沉积、胶质细胞增生等。
临床特点:①该病多见于儿童青少年,以 5~15岁最好发,也可见于成人,本例患者是55岁中年女性;②临床表现复杂多样,缺乏特异性,多以间断性头痛、呕吐、智力障碍、肌力异常、癫痫发作等为首发症状,且症状进行性加重;③血和脑脊液检查,乳酸和丙酮酸增高,本例血和脑脊液乳酸检查均升高;④脑电图、肌电图检查,表现异常,呈癫痫表现,肌源性或神经源性损害;⑤肌肉活检:可见线粒体异常和不整红边纤维(RRF);⑥基因检测:mtDNA中tRNA亮氨酸(Leu)基因核甘4243位点发生A-G点突变。
MRI表现:①发病部位及病变形态,好发于顶枕叶、颞叶的脑皮层及皮层下区,病变分布与脑动脉供血区不一致;病变形态不规则呈斑片、条状,病变区灰白质界限欠清楚;②信号特点:呈长T1长T2信号,FLAIR及DWI 均呈高信号,ADC值降低, PWI示病变区呈高灌注状态;③病灶数目:单发或多发对称性病灶,可新旧病灶并存,具有此起彼伏、游走的特点;④局部脑萎缩:陈旧病灶区脑皮质变薄,脑沟裂增宽,呈萎缩样改变;⑤增强扫描:病灶无强化,或轻度脑回样强化;⑥MRS:具有特征性,病变区表现NAA降低和Lac升高, NAA/Cr降低,Lac/Cr升高;另外,脑脊液区1H-MRS也可出现Lac峰。周志凌等[1]报道13例线粒体脑病中10例患者脑脊液区可见Lac峰,有助于对脑脊液乳酸水平的检测;⑦MRA:表现大脑中、后动脉分支的小血管增多,但罕有大血管闭塞或狭窄。
鉴别诊断:线粒体脑肌病的病变形态和信号与脑梗死和脑炎[2]相似,容易混淆需要鉴别。①急性脑梗死:发病急,病变部位与脑血管供血范围一致,脑内血管多有不同程度血管闭塞或狭窄改变,增强扫描常有明显脑回样强化;而线粒体脑肌病病变部位与血管供应范围不一致、且多发病变常不在同一血管支配区,病变可在短期内好转或进展,具有多灶性、对称性、游走的特性,增强扫描病灶多不强化或仅见轻度沿病变脑回分布的线样强化。②脑炎:临床常有发热、头痛,甚至昏迷等症状,脑脊液检查也具有其表现特点,患者多有感染史,主要累及颞叶和边缘系统。
