用于氢、电转换与存储的功能性硼氢化物
2022-05-13李文晶任开响李永涛张庆安
李 晨,李文晶,任开响,李永涛,张庆安
(1.安徽工业大学材料科学与工程学院,安徽 马鞍山 243002)(2.国家石油储备中心,北京 100045)
1 前 言
由于化石燃料的有限供应及其对气候和环境的不利影响,开发清洁、可持续的替代能源成为全球共识[1]。为满足世界能源的重大需求,太阳能、风能、电能和氢能利用技术应运而生;其中,氢能和电能作为二次能源,因其具有多样化的来源、转换及利用方式而备受关注。然而,如何实现氢/电能的安全、高效储存与转换成为当前面临的主要技术挑战[2]。
配位硼氢化物属于含共价键的离子化合物,易发生多晶型转变,其高温离子迁移率远大于常规聚合物电解质,同时具有较高含氢量,因而备受关注。在很长一段时间内,硼氢化物被作为化学合成还原剂和火箭燃料来使用[2, 3]。随着人们发现硼氢化物还是一种良好的氢载体和离子导体,其被广泛用于固态储氢、水解制氢、全固态电池等领域[4-6]。最近,作者团队[7]发现硼氢化物还可用于提高钙钛矿太阳能电池(perovskite solar cells,PSCs)的光电转换效率和稳定性。上述研究表明,配位硼氢化物作为载体在能源转换与存储过程中发挥了多种功能性作用。
为此,本文概述了配位硼氢化物的功能性应用,包括其在固态储氢、水解制氢、全固态电池、太阳能电池领域的最新进展,分析了其存在问题及可能的解决路径,并探讨了其未来的发展趋势。
2 固态储氢
氢能因其来源丰富、燃烧效率高、无污染等特点,被认为是绿色、可持续的二次能源。然而,如何实现高效、安全存储与输运成为制约其规模化应用的难题[2]。相比于高压气态储氢和低温液态储氢,基于轻金属硼氢化物的固态储氢展现出明显的优势,包括储氢密度高、安全可靠、使用效率高等优点,被认为是目前最具潜力的储氢方式[4]。
代表性的轻金属硼氢化物包含LiBH4、NaBH4、Ca(BH4)2、Mg(BH4)2等[8-11]。金属硼氢化物由B—H共价键和金属离子键作用形成,热力学稳定性高,一般也具有较高的放氢温度,其分解反应方程式如下:
MBH4↔MH+B+3/2H2M=Li, Na,Mg
(1)
图1给出了常见的碱金属或碱土金属硼氢化物、锆硼氢化物和铝硼氢化物的体积储氢密度、质量储氢密度和分解温度(decomposition temerprature,Td)[12],由图可知,LiBH4的质量储氢密度最大,但Td也较高[7],大部分氢在400 ℃以上才能缓慢释放。因此,如何通过调控材料组成、结构和反应过程,使其在温和条件下实现可逆吸/放氢,成为当前研究的重点。目前已报道的性能改善策略有纳米调制[11]、离子替代[13]、催化掺杂[14]、反应失稳[15]等。其中,纳米调制策略在降低材料热稳定性和提高材料可逆加氢方面都展现出巨大优势。因此,本节将重点介绍纳米调制改善硼氢化物储氢性能的最新进展。
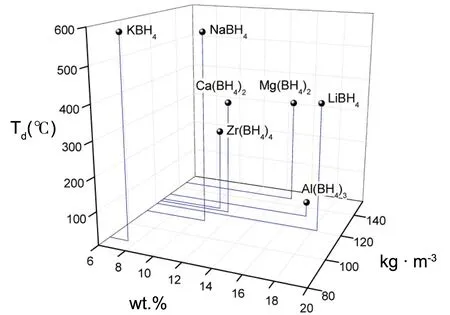
图1 金属硼氢化物的质量储氢密度、体积储氢密度和分解温度[12]Fig.1 Weight hydrogen density, volume hydrogen density and decomposition temerprature of metal borohydrides[12]
研究表明,可以通过减小粒子尺寸形成纳米颗粒改变硼氢化物动力学和热力学性能[16]。通过机械球磨法和溶液蒸发法制备的纳米LiBH4颗粒,可以明显提高其放氢动力学性能[17]。此外,通过浸渍法将LiBH4装载于SBA-15、C60、碳气凝胶、有序多孔碳(CMK-3)、活性炭或碳纳米管等载体中,可以借助纳米尺寸效应来提高复合材料储氢性能[18-21];同时纳米介孔支架还可以用来限制LiBH4粒子的长大和阻止循环吸/放氢过程中的纳米颗粒团聚现象[20]。Xu等[22]通过溶液浸渍法成功地将LiBH4限制在分级多孔ZnO/ZnCo2O4(ZZCO)纳米颗粒中。通过纳米约束和ZZCO失稳协同作用可使该复合材料获得优异的放氢性能。LiBH4@ZZCO样品可在169 ℃时释放氢气,最大放氢峰位于275 ℃,反应激活能(Ea)降低至120 kJ·mol-1,远低于纯LiBH4。
Suwarno等[20]利用准弹性中子散射和量热测量研究纳米多孔二氧化硅支架和碳基支架对LiBH4的约束效应,二氧化硅孔道半径可从20 nm缩小至0.8 nm,孔道尺寸越小,纳米约束效应越明显。与孔径尺寸作用类似,硅基支架的约束效应比碳基支架更明显。准弹性中子散射结果证实了硅基材料中LiBH4的氢迁移率高于碳基材料中的。此外,靠近孔壁的LiBH4非晶层表现出与结晶LiBH4截然不同的相行为。这些结果表明,除了孔径大小外,支架的化学性质也对纳米调制约束硼氢化物的氢迁移率和界面层厚度起着重要作用。
浙江大学肖学章等[11]将LiBH4装载于高度有序的介孔碳(MC)支架上,并且将NbF5纳米颗粒均匀分散在基体上。纳米LiBH4@MC-NbF5体系的初始放氢温度为150 ℃,相比于块状LiBH4降低了225 ℃,改善效果显著;其放氢活化能从块状LiBH4的189 kJ·mol-1降低到98 kJ·mol-1;吸氢温度降低到温和的200 ℃,6 MPa H2。上述材料性能的提高归因于Nb基催化和LiBH4颗粒纳米化协同效应,这为提高硼氢化物的可逆储氢性能提供了一个重要的方法。
针对NaBH4的放氢温度非常高(>500 ℃),而且无法在温和的条件下进行可逆吸/放氢反应的问题,安徽工业大学李永涛等[23]通过机械力诱导自组装策略,以工业石墨为碳载体来源,借助金属硼氢化物与石墨材料之间的内在相互作用,通过原位形成的硼氢化物细小颗粒的插入和吸附,同步实现了硼氢化物诱发石墨固态剥离以及石墨片指引硼氢化物颗粒形貌重构,成功获得了石墨纳米片负载的超细金属硼氢化物纳米点。如图2a~2g所示,均一的NaBH4纳米点附着在石墨片层上,标记为nano-NaBH4@GNs。从图2h~2m中可得出:① 复合材料热力学性能得到显著改善,如490 ℃时放氢平台压力由micro-NaBH4的0.025 MPa升高到nano-NaBH4@GNs的0.18 MPa,对应的氢气分解焓变由micro-NaBH4的(102.9±3.6) kJ·mol-1降至nano-NaBH4@GNs的(56.5±0.5) kJ·mol-1。② 复合材料放氢动力学明显提升,在500 ℃时,nano-NaBH4@GNs在120 min释放~6.8%氢气(质量分数,下同),而micro-NaBH4在300 min仅释放~1%;即使在更低温度下,nano-NaBH4@GNs依然具有较好性能,如其在450和400 ℃时,300 min内分别释放~5.2%和~4.1%氢气;而在相同400 ℃条件下,micro-NaBH4几乎不放氢。动力学定量计算表明,nano-NaBH4@GNs的放氢激活能为(41.3±4.7) kJ·mol-1,相对于所报道氟化物掺杂的micro-NaBH4降低了~80%(Ea≈220 kJ·mol-1)。上述结果表明,相比于micro-NaBH4,nano-NaBH4@GNs的放氢热力学和动力学性能均得到显著改善。该研究不仅实现了金属硼氢化物纳米晶的快速、简易制备,而且还阐明了纳米晶的形成机理和可逆储氢改善机制。
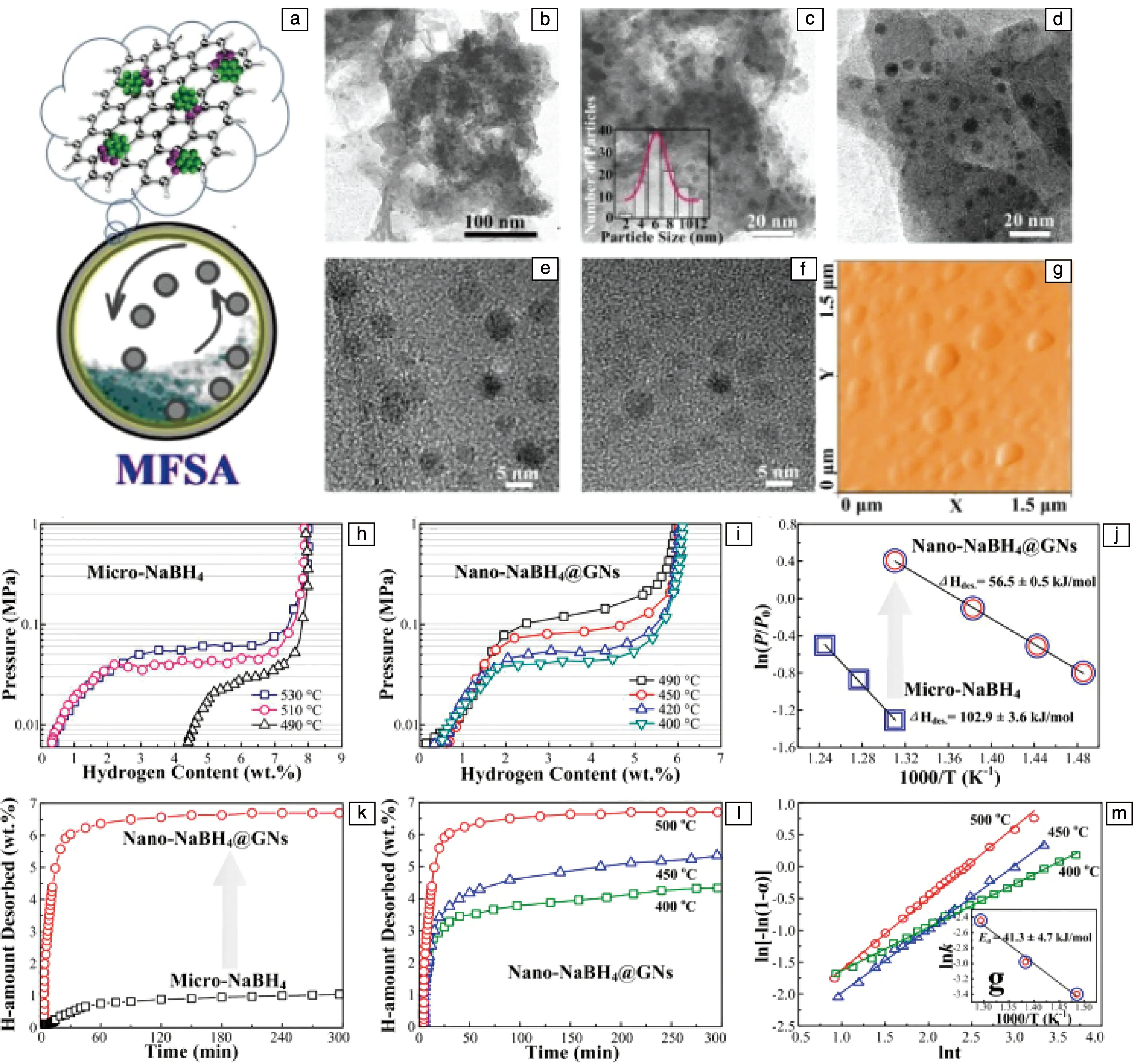
图2 采用机械力诱导自组装策略制备石墨纳米片原位负载NaBH4纳米点复合材料(nano-NaBH4@GNs)示意图(a),nano-NaBH4@GNs的TEM照片、粒径分布图、HRTEM图片和AFM照片(b~g);nano-NaBH4@GNs和micro-NaBH4的放氢热力学和动力学性能比较:(h,i)不同温度放氢P-C曲线,(j)Van’t Hoff拟合曲线,(k)500 ℃等温放氢曲线;不同温度下nano-NaBH4@GNs等温放氢曲线(l)和JMA模型拟合曲线(m)[23]Fig.2 Schematic illustration of the synthesis of NaBH4 nanodots on graphene (nano-NaBH4@GNs) by mechanical force induced self-assembly strategy (a), TEM image, grain size distribution, HRTEM image and AFM images of nano-NaBH4@GNs (b~g); comparison of hydrogen desorption thermodynamic and kinetic properties between nano-NaBH4@GNs and micro-NaBH4: (h, i) P-C desorption isotherms at different temperatures, (j) Van’t Hoff fitted curves, (k) isothermal hydrogen desorption curves at 500 ℃; isothermal hydrogen desorption curves (l) and JMA model fitting curves (m) for the nano-NaBH4@GNs at differrent temperatures[23]
复旦大学余学斌等[24]通过在MgH2表面构建异质结构来改善Mg(BH4)2的可逆储氢性能。如图3所示,以石墨烯负载的平均粒径约为10 nm的MgH2颗粒为反应基体,通过其与B2H6发生原位固相反应,在颗粒表面构建壳层,形成Mg(BH4)2@MgH2核-壳纳米结构。进一步吸/放氢性能检测结果表明,MgH2和Mg(BH4)2的放氢温度都随颗粒尺度减小而降低。通过构建Mg(BH4)2@MgH2纳米颗粒(记为MBH@MH-2),所含MgH2和Mg(BH4)2的Td分别降低到~191和~242 ℃。特别是在MgH2作用下,所制备的Mg(BH4)2的可逆循环效率可以达到68%,而纯Mg(BH4)2可逆效率仅有34%。理论计算结果证明,在MBH@MH-2异质结构中,MgH2的H原子优先与MgB2中的B原子发生键合作用,使得部分B—B键自发断裂而生成B—H键,这在热力学方面有利于Mg(BH4)2的可逆加氢性能。
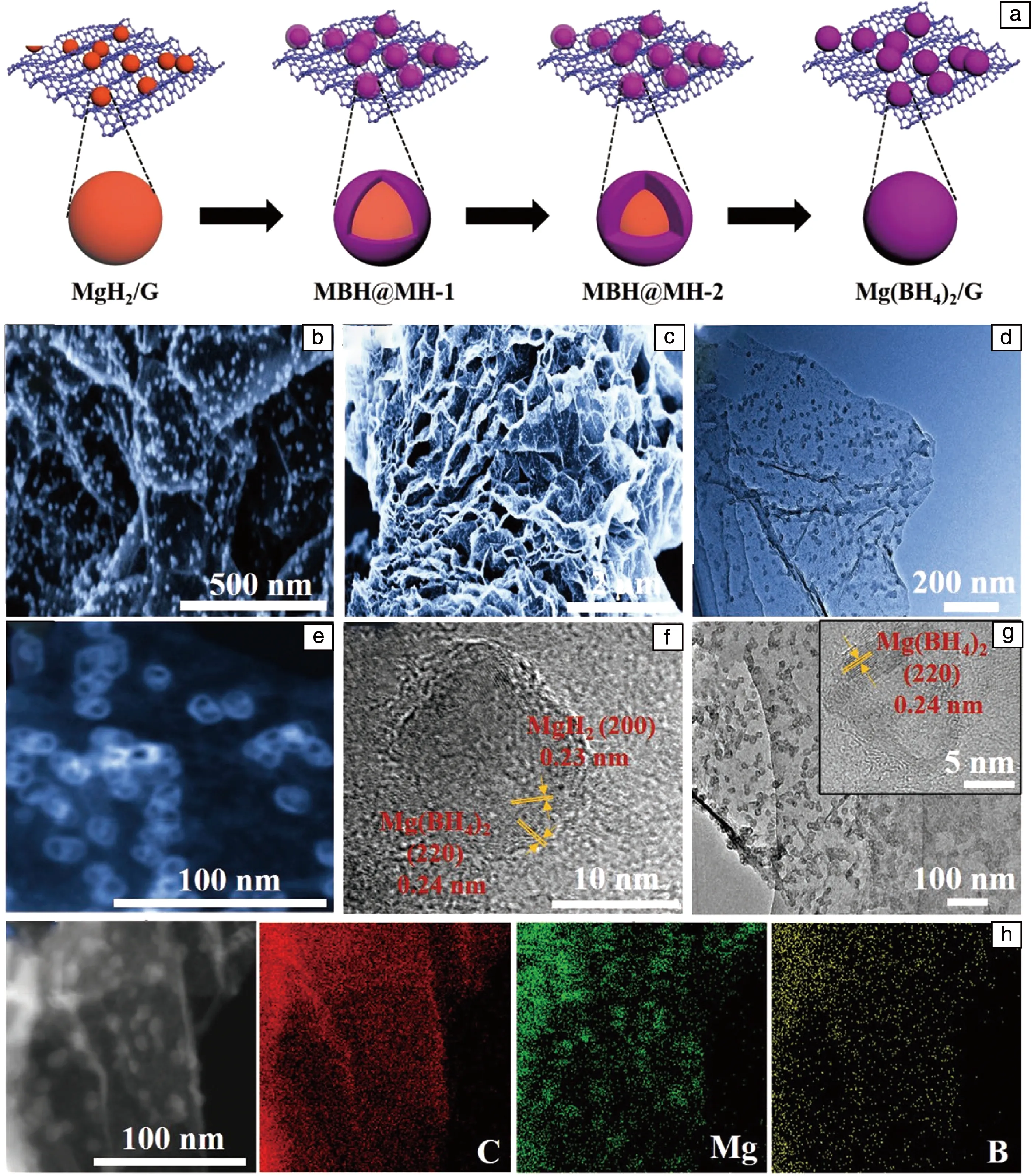
图3 石墨烯负载Mg(BH4)2@MgH2纳米颗粒(Mg(BH4)2/G):(a)制备流程示意图,(b,c)SEM照片,(d)TEM照片;Mg(BH4)2@MgH2(MBH@MH-2)样品的STEM(e)和HRTEM(f)照片;Mg(BH4)2/G样品的TEM照片(g),MBH@MH-2样品的元素面分布图(h)[24]Fig.3 Mg(BH4)2@MgH2 nanoparticles on graphene (Mg(BH4)2/G): (a) schematic illustration of the synthesis process, (b, c) SEM images, (d) TEM image; STEM (e) and HRTEM (f) images of Mg(BH4)2@MgH2 (MBH@MH-2); TEM image of Mg(BH4)2/G (g); Elemental mapping of MBH@MH-2 (h)[24]
浙江大学刘永锋等[25]基于一锅溶剂热方法,如图4a所示,利用丁基锂、三乙胺硼烷和二茂镍为反应前驱体,生成了石墨烯负载、原位纳米Ni催化的超细LiBH4纳米颗粒复合储氢材料(nano-LiBH4/Ni@G),其中负载的LiBH4的含量高达70%。进一步对该复合材料微观结构和形貌的分析表明,石墨烯表面均匀分布着5~10 nm的LiBH4和3 nm左右的Ni纳米颗粒,两者有明显的交叠现象。上述材料借助纳米效应和Ni催化协同来改善LiBH4的可逆储氢性能,实现在较温和的条件下可逆储存9.2%的氢气(如图4b)。经过70个循环后,nano-LiBH4/Ni@G样品微观结构和储氢容量保持了较高的稳定性(图4c~4f)。该合成策略可扩展到其他配位氢化物的纳米化制备,有望推动固态储氢材料的实用化发展。
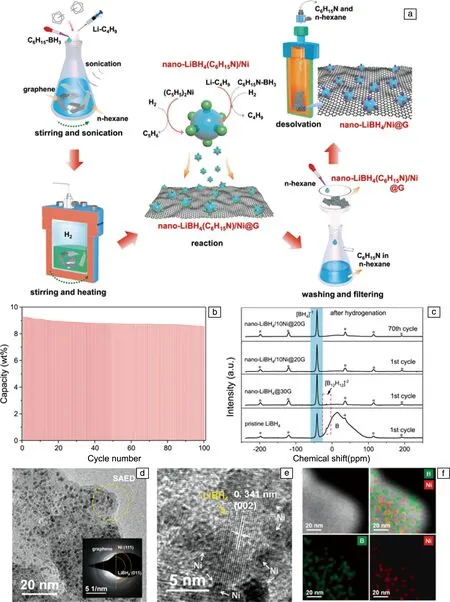
图4 Nano-LiBH4/Ni@G复合物:(a)制备流程示意图,(b)吸放氢循环稳定性,(c)11B固态NMR谱图;nano-LiBH4/10Ni@20G循环70次后:(d)TEM照片,(e)HRTEM照片,(f)EDS元素面分布图.图4c中*表示旋转边带,图4d中插图为SAED衍射花样[25]Fig.4 Nano-LiBH4/Ni@G composite: (a) schematic illustration of preparation process, (b) hydrogen cycling stability, (c) solid state 11B NMR spectra; nano-LiBH4/10Ni@20G after 70 cycles: (d) TEM image, (e) HRTEM image, (f) EDS mapping. The peaks marked with*in Fig.4c indicate the spinning sidebands and the insert in Fig.4d is the SAED pattern[25]
综上所述,纳米结构调制能明显降低金属硼氢化物热力学稳定性,提高动力学性能和可逆性,成为调控该类材料储氢性能的重要手段。然而,固态储氢材料离实用化还较远,因此,结合其他有效方法建立协同策略改善固态储氢材料性能是未来发展的主要趋势,尤其是纳米调制与催化掺杂、反应失稳体系构建相结合策略。
3 水解制氢
水解制氢主要是通过硼氢化物与水发生反应释放氢气,故通常为不可逆反应。常见的水解制氢材料主要包含金属、配位氢化物以及氨硼烷等[26]。其中,配位氢化物NaBH4因其理论贮氢量高(10.8%)、在碱性溶液中稳定性好、无毒、产氢纯度高而备受关注[27, 28]。本节主要从水解反应、催化剂和可逆再生方面介绍NaBH4基水解制氢。
NaBH4水解制氢主要通过以下反应进行:
NaBH4+2H2O→NaBO2+4H2
(2)
然而,NaBH4在水中的溶解度较低,室温下低于0.16 g·g-1,仅部分溶于水的NaBH4发生水解反应,导致实际氢气产率较低,且产物为NaBO2·xH2O[29]。因此,其本质水解过程为:
NaBH4+(2+x)H2O→NaBO2·xH2O+4H2
(3)
如何提高氢气生成率和实现水解副产物(NaBO2)的低成本再生成为商业化应用的技术挑战。使用合适的催化剂成为提高NaBH4产氢反应速率的主要策略。目前已有的催化剂主要包括无机酸(HCl、HNO3、H2SO4)、羧酸(HCOOH、CH3COOH)、贵金属单质、金属卤化物和硼化物等[30, 31]。其中,贵金属具有高的催化活性和化学稳定性,但较高的成本限制了其规模化应用。
开发非贵金属及化合物成为新型催化剂设计的方向。Zhang等[32]以CoSO4为前驱体,制备了掺杂CoSO4的Co3O4样品,该样品表现出优异的水解催化活性。进一步向上述样品中分别引入Na2SO4或Na2S,将有利于Co3O4原位转化为具有催化活性的Co-B合金;与Na2SO4相比,Na2S水解过程还引起了Co-B合金表面形态发生变化,使得氢气的释放非常迅速,在5 min内达到300~450 mL(图5a~5c),且氢气生成速率提高至4425 mL·min-1·g-1。
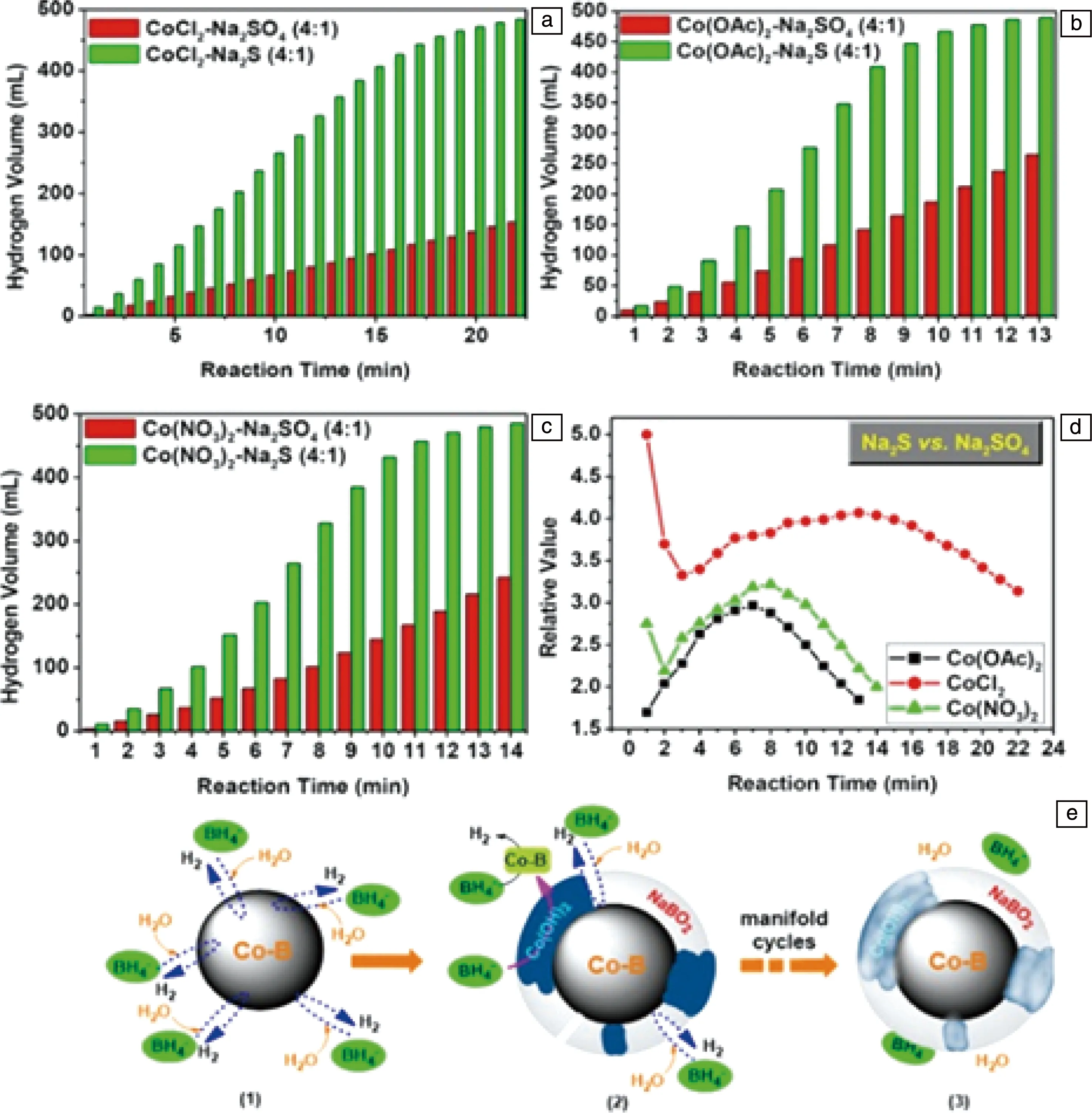
图5 分别添加Na2SO4与Na2S对无硫Co基催化剂活性的影响:(a)CoCl2基催化剂,(b)Co(OAc)2基催化剂,(c)Co(NO3)2基催化剂;添加Na2SO4或Na2S对无硫Co基催化剂水解反应速率的影响(d)及其水解示意图(e)[32]Fig.5 Contrast effect of Na2SO4 and Na2S on the reactivity of those catalysts derived from sulfur-free cobalt sources: (a) CoCl2-made catalyst, (b) Co(OAc)2-made catalyst, (c) Co(NO3)2-made catalyst; effect of the hydrolysis reaction rate (d) and their Sketch maps (e) of those Co-based catalyst[32]
此外,在钴基催化剂上分别添加上述含硫盐,使其在加速制氢反应中表现出不同的催化反应速率(图5d),其中,添加Na2SO4和Na2S对Co3O4-CoCl2催化剂的催化速率有明显的增强,有效地提高了Co基催化剂的制氢活性。近期Lai等[33]制备了Ni掺杂NaBH4纳米核壳结构复合材料,发现该复合材料在10~60 ℃或水过量情况下,均呈现出较高氢生成率,如60 ℃时产氢速率达到22.5 g·min-1;其水解副产物为NaB(OH)4,可通过化学途径回收,重新生成NaBH4。
对于水解副产物再生方面,研究人员也作了大量的尝试。华南理工大学Ouyang等[34]报道了无需引入氢化物或氢气的水解副产物简易、廉价的再生方案。如图6所示,通过在室温下球磨Mg和NaBO2·2H2O(或NaBO2·4H2O)即可获得再生NaBH4,再生NaBH4中的H+源自于水解副产物所带的吸附H2O,该再生方案的产率高达68.55%。在CO2气氛下球磨水解产物NaBO2·2H2O和Mg同样可以实现NaBH4的再生。水溶液中的水解产物NaBO2与CO2反应形成Na2B4O7·10H2O和Na2CO3,将两者在室温条件下与Mg一起球磨可形成NaBH4,该方法可使水解副产物再生率提升至80%。上述再生路径不仅避免了昂贵的还原剂(如Mg(OH)2)和高能耗的脱水过程,还无需高压H2,因此大大降低了成本。
这话王爷听在耳朵里很多次。他不言语,只笑笑。一个女人,不嫌自己掏厕所身上臭,不嫌公婆病了她瘦弱的身子独自拖架子车送到十几公里远的镇卫生站,更不嫌他病退后不另想办法多挣点钱成天务弄这些个没用的,应该算是王爷他自己得着了大便宜,这样的女人如今到哪里找,谁又能替代得了呢。
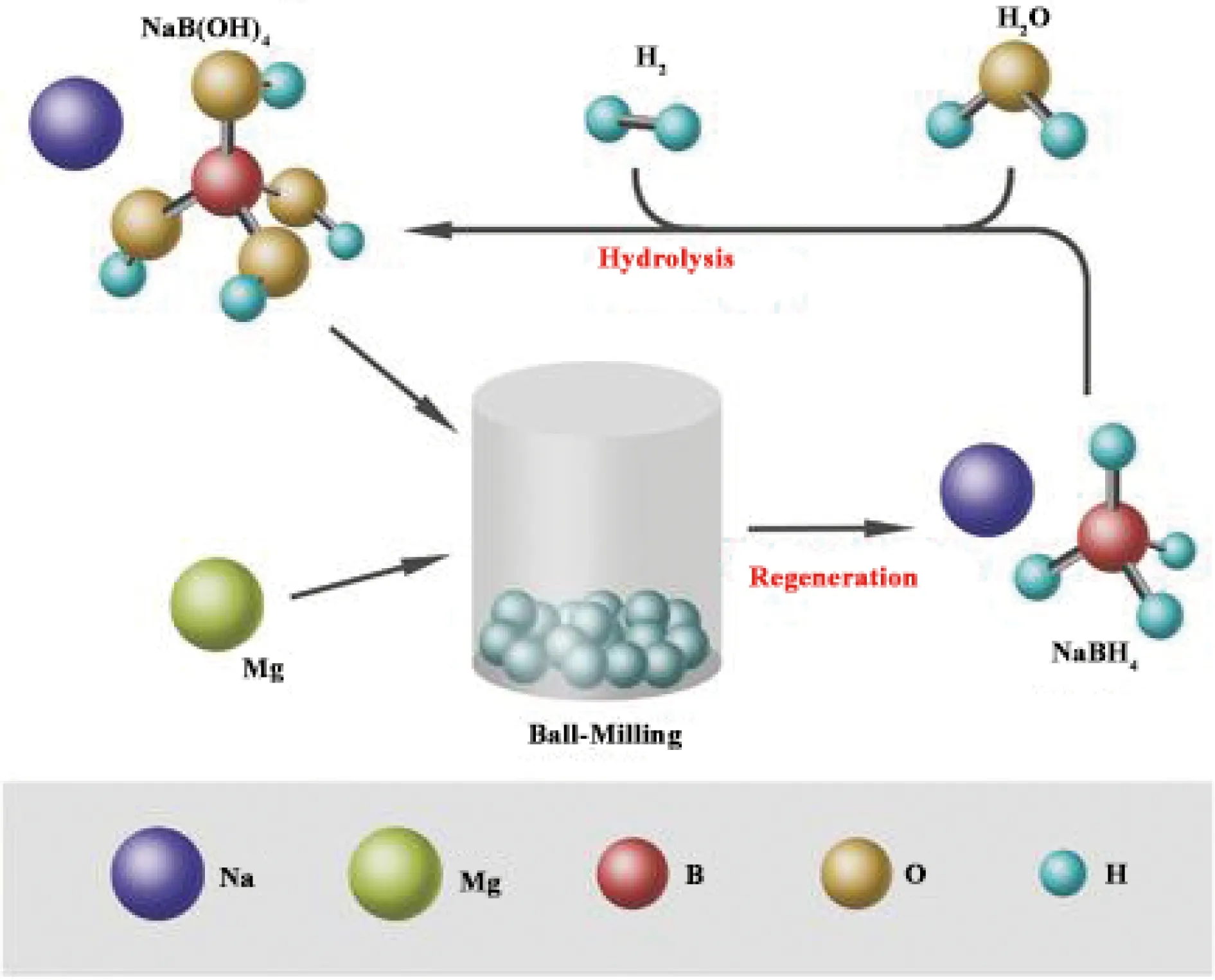
图6 水解副产物NaBO2·2H2O再生NaBH4的制备原理图[34]Fig.6 Preparation schematic diagram of regenerated NaBH4 by hydrolysis byproduct NaBO2·2H2O[34]
总之,在硼氢化物水解制氢应用方面,开发非金属催化剂已取得突破,实现了低成本、可控制氢,目前研究的重点集中于副产物的可逆再生方面。
4 全固态电池
相对于通常采用有机液体或凝胶作为电解质的传统锂离子电池,采用固体电解质的全固态电池具有更高的能量密度、功率密度和安全性[35]。然而,阻碍固体电解质规模化应用的最大技术挑战在于如何在保持其电化学稳定性的同时还能使其具有较高的离子电导率[36]。因此,开发高电导率的新型固体电解质材料成为当前研究的热点。
配位硼氢化物LiBH4包含金属阳离子Li+和配位阴离子[BH4]-,具有明显的离子晶体特性。如图7所示,LiBH4室温下以斜方Pnma相存在,离子电导率很低,约为10-5mS·cm-1;在118 ℃时,其晶型转变为高温六方P63/mmc结构[37-39],离子电导率得到显著提升,可达到1 mS·cm-1。基于此,2009年,Orimo等[40]首次将高温型LiBH4用作固体超离子导体。然而,从实用固体电解质出发,如何使LiBH4在近室温下仍保持较高的离子电导率成为技术挑战。下面将从4大改善路径来概述相关最新研究进展。
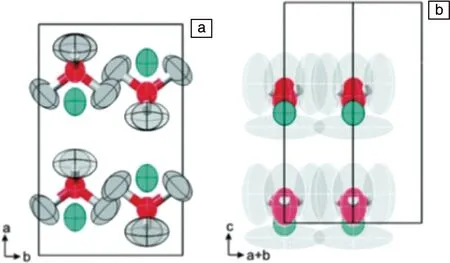
图7 LiBH4的晶胞示意图[39]:(a)低温Pnma相结构,(b)高温P63/mmc相结构.灰色:H原子,红色:B原子,绿色:Li原子Fig.7 Cell structure diagram of LiBH4[39]: (a) low-temperature Pnma structure, (b) high-temperature P63/mmc structure. Grey: hydrogen atom, red: boron atom, green: lithium atom
4.1 卤元素替代
通过卤元素替代来提高LiBH4电解质使用温度下的离子电导率,其内在本质是使高温相在低于相变点温度、甚至室温下仍然稳定存在[38]。
Gulino等[41]在室温下采用卤离子取代部分络合阴离子[BH4]-来稳定LiBH4的六边形结构,首次在室温下得到了含Cl-和Br-卤化物的稳定六方固溶体LiBH4-LiBr-LiCl。在相同的温度范围内,固溶体LiBH4-LiBr-LiCl的离子电导率值都大于纯LiBH4。其中,h-Li(BH4)0.7(Br)0.2(Cl)0.1的电导率最高(30 ℃时为1.3×10-5S·cm-1),比纯LiBH4的离子电导率(30 ℃时为10-8S·cm-1)高约3个数量级。Nguyen等[42]和Ley等[43]采用球磨法制备了LiM(BH4)3Cl(M=La,Ce,Gd)化合物,该化合物都具有良好的离子电导率,室温下离子电导率超过1×10-4S·cm-1,70 ℃时离子电导率约为1×10-3S·cm-1;而且电子电导率也被控制在极低水平(<10-7S·cm-1)。
4.2 纳米限域
纳米限域将LiBH4熔铸到多孔纳米结构内,可以有效降低LiBH4的相转变温度点,甚至将其相转变温度降低至室温,较高的Li+流动性使得Li+电导率得到大幅度提升[44, 45]。此外,材料与支架作用还能形成新的纳米界面,从而有利于离子迁移,改善材料的电化学性能[46]。
研究发现,介孔二氧化硅MCM-41负载LiBH4纳米复合物由于LiBH4和二氧化硅之间形成界面层,有利于Li+迁移,而使得该复合物具有较高的离子电导率,尤其使其低温时也具有很高的电导率[47]。最近,上海理工大学郑时有课题组[48]将LiI掺杂的LiBH4约束在介孔二氧化硅SBA-15中,合成了具有高Li+导电性的物质Li4(BH4)3I@SBA-15。如图8a所示,相比于LiBH4的离子电导率在105 ℃时出现了3个数量级的突然下降,Li4(BH4)3I@SBA-15在105 ℃时电导率突然下降的现象消失,其对数电导率随温度的倒数(1/T)增加而线性下降,这些现象表明LiBH4的高温P63/mmc相通过I-取代可稳定到室温存在。Li4(BH4)3I@SBA-15离子电导率在125 ℃时达到1.0×10-2S·cm-1,在35 ℃时达到2.5×10-4S·cm-1,几乎比LiBH4提高了4个数量级。这些结果表明,I-取代和纳米约束的协同作用改善了LiBH4的电导率。如图8b和8c所示,使用直流极化和电化学阻抗技术测量35 ℃时Mo|Li4(BH4)3I@SBA-15|Mo电池在10 mV时的稳定电流非常小,表明Li4(BH4)3I@SBA-15的电子转移数接近0。进一步根据Li|Li4(BH4)3I@SBA-15|Li电池在10 mV的初始稳定电流和极化前后的电阻,确定了Li+迁移数为0.97。上述结果证实Li4(BH4)3I@SBA-15中大部分的电荷转移可通过Li+迁移来实现。

图8 LiBH4、Li4(BH4)3I、LiBH4@SBA-15和Li4(BH4)3I@SBA-15的温度依赖性离子电导率(a);通过直流极化(b)和电化学阻抗技术(c)测量Li4(BH4)3I@SBA-15的电子迁移数[48]Fig.8 Temperature-dependent conductivities of LiBH4, Li4(BH4)3I, LiBH4@SBA-15 and Li4(BH4)3I@SBA-15 (a); transference number measurements of Li4(BH4)3I@SBA-15 by direct current polarization (b) and electrochemical impedance spectroscopy technique (c)[48]
4.3 聚阴离子构筑
闭合型配位硼氢化物中聚阴离子的硼和氢原子具有独特的共价键模式,形成了坚固稳定的笼型聚阴离子结构,如图9所示[49]。Orimo等[50]首次在LiBH4放氢过程中发现中间相产物Li2B12H12,之后在NaBH4、Mg(BH4)2和Ca(BH4)2的分解反应中又发现了Na2B12H12、MgB12H12和CaB12H12[51-53]。十二硼氢化物M2/nB12H12(n代表金属M的价态)中包括二十面体阴离子团[B12H12]2-,其具有超高的电导率、较高的热力学和化学稳定性,可以作为电解质应用在固态电池中。
Udovic等[55]报道了一种闭合型十氢十硼酸钠(Na2B10H10),其同样具有较大的椭圆形B10H102-阴离子(图9),在约87 ℃以上转变成无序的面心立方(fcc)相,且具有富含空位的Na+阳离子亚晶格。这种阳离子亚晶格在由B10H102-阴离子形成的宽敞通道内具有高度的流动性,并表现出显著的超离子导电性,在110 ℃时Na+电导率为0.01 S·cm-1,相同温度下,其Na+电导率明显高于Na2B12H12[56]。
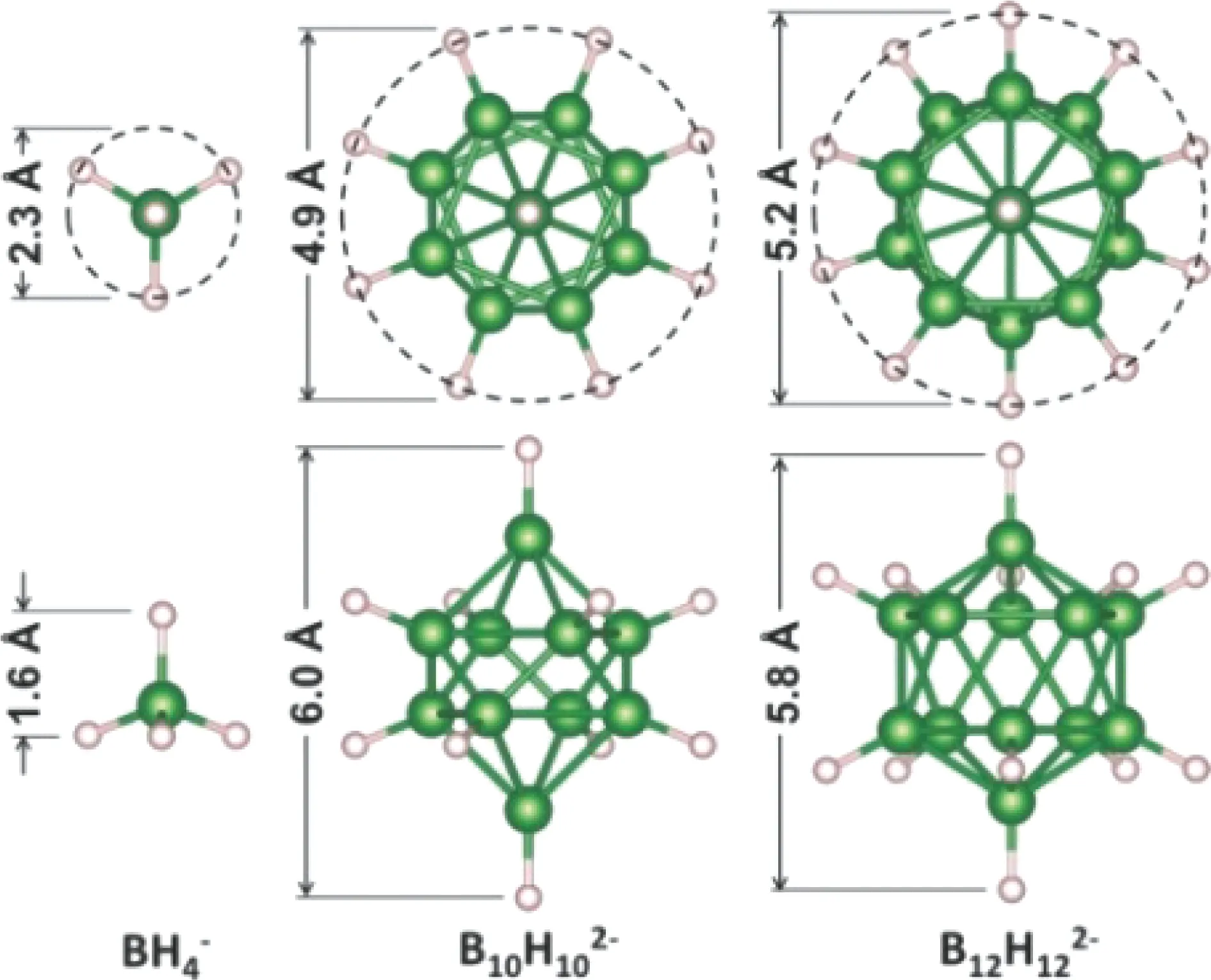
图9 分别从俯视和侧视角度显示的BH4-、B10H102-和B12H122-阴离子的晶胞结构和几何尺寸,硼原子和氢原子分别用绿色和粉色的球体表示[49, 55]Fig.9 Geometries and approximate relative sizes of BH4-, B10H102- and B12H122- anions, each shown from top and side views, the boron and hydrogen atoms are denoted by the green and pink spheres, respectively[49, 55]
在上述研究的基础上,有研究通过引入另一种离子形成双金属阳离子或者双阴离子十二硼氢化物来提高硼氢化物的离子电导率[57]。Sadikin等[58]提出了新型固体电解质Na3BH4B12H12和(Li0.7Na0.3)3BH4B12H12,该电解质均由LiBH4、NaBH4、Li2B12H12或Na2B12H12其中2种混合组成。含有硼氢化物和闭合型硼烷相的复合物在室温下显示出比Na2B12H12更高的电导率值。当温度升高时,所有复合混合物的电导率显著增加,这归因于其形成了混合的闭合型硼烷相。所形成的Na3BH4B12H12在室温下电导率约为5×10-4S·cm-1,比其他钠导电硼氢化物材料离子电导率值,如Na2BH4NH2离子电导率(2×10-6S·cm-1),高2个数量级以上[59]。最近,Tang等[49]报道了碳掺杂聚阴离子化合物MCB9H10(M=Li,Na),该化合物含有类似[B10H10]2-的单价、双环-笼形CB9H10-阴离子。其中,接近室温下LiCB9H10和NaCB9H10的离子电导率均大于0.03 S·cm-1。构筑多面体聚阴离子策略使硼氢化物有望成为下一代固体电解质。
4.4 多元复合
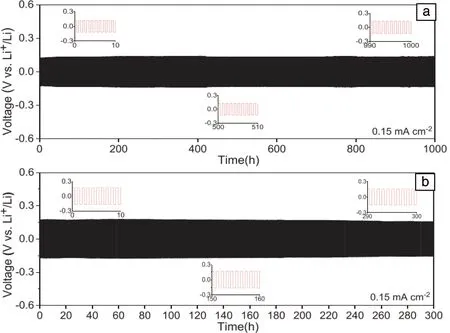
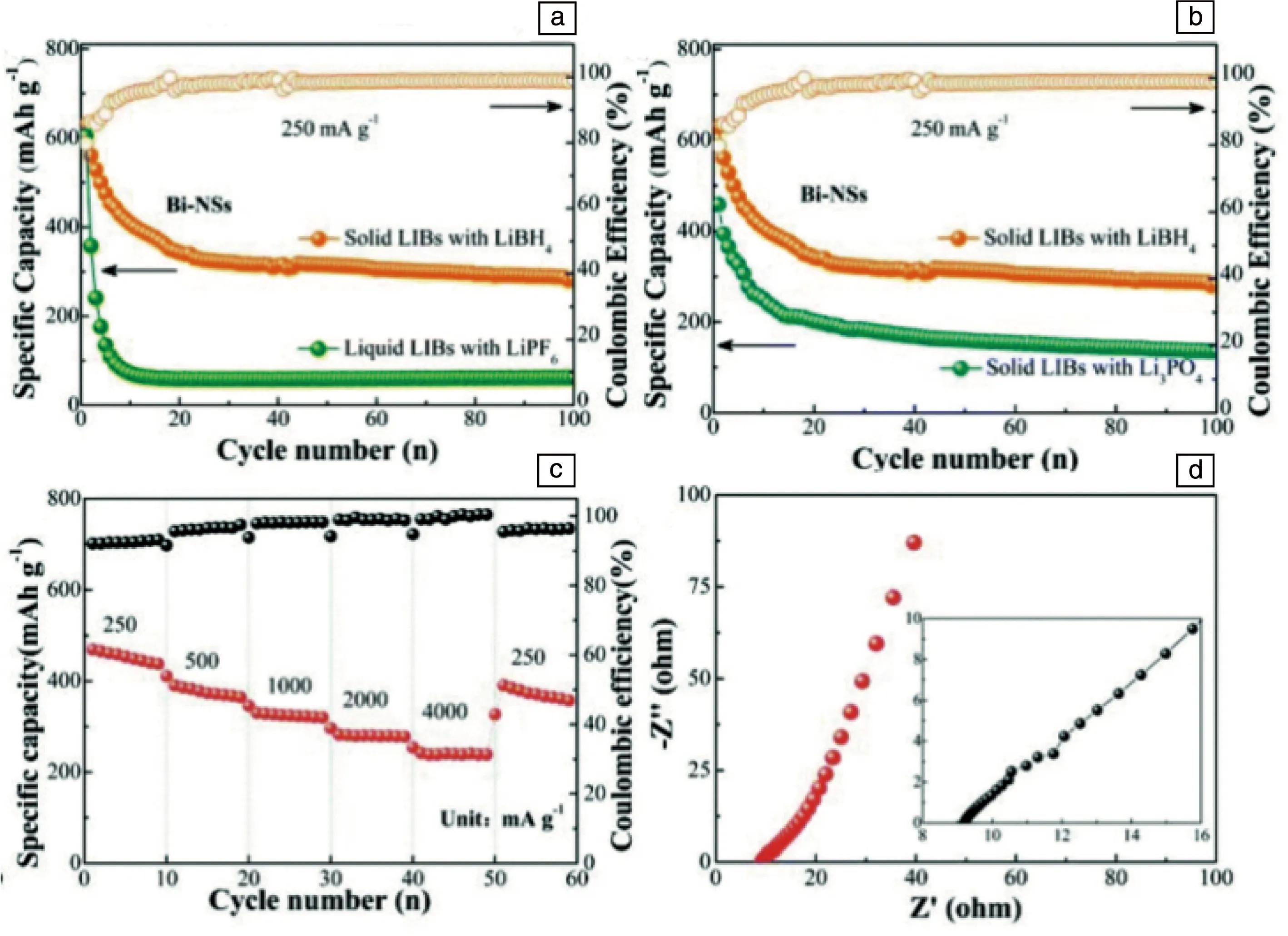
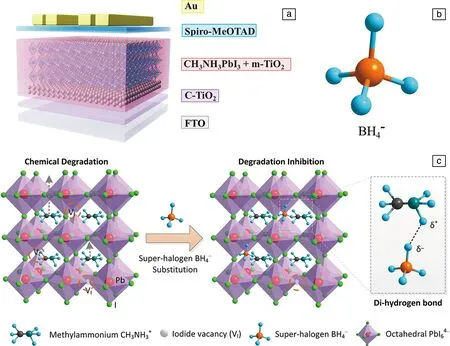
Zhang等[60]提出了通过氨合反应来提高LiBH4的离子电导率,并证明了硼氢化锂氨化物在室温下通过氨气(NH3)的吸收和解吸来实现自身离子电导率的调控。将LiBH4置于NH3气氛中加热可形成结晶度良好的Li(NH3)BH4,其室温下Li+电导率高达10-3S·cm-1。随着温度升高,其晶体结构由Li(NH3)BH4逐渐转变为Li(NH3)xBH4(0.5 Liu等[61]报道了一种新型的室温超快Li+导体——硼氢化锂氨硼烷配合物((LiBH4)x·NH3BH3)。研究表明,将NH3BH3加入到LiBH4结构中引起晶胞体积增加,降低了Li+的体积密度,从而有利于Li+迁移。在25 ℃时,LiBH4·NH3BH3复合物的离子电导率高达4.0×10-4S·cm-1,而电子电导率几乎可以忽略不计;此外,该超快Li+导体在18~55 ℃时都能保持稳定。 Gao等[62]采用高能球磨法成功地制备了内、外层分别为LiBO2和LiBH4的双层非晶涂层Li6.4La3Zr1.4Ta0.6O12(LLZTO)复合材料。之后将LiBH4与LLZTO颗粒复合来获得具有高离子电导率和良好电化学性能的复合材料。如表1所示,LLZTO-xLiBH4复合材料的离子电导率随LiBH4含量的变化呈现先增加后降低的趋势。在30 ℃时该复合材料Li+电导率最大,约为8.02×10-5S·cm-1,比未烧结的LLZTO的电导率(4.17×10-9S·cm-1)提高了4个数量级,甚至接近高温烧结制备的LLZTO的Li+电导率[47]。 表1 LLZTO-xLiBH4样品的离子电导率和活化能[62] 图10a和10b分别给出了60和30 ℃时Li|LLZTO-4LiBH4|Li的恒电流循环曲线。在60 ℃时,其循环时间超过1000 h,无明显的电压波动,过电位为130 mV;在30 ℃循环,其过电位略微增加到150 mV,循环300 h后电压仍然保持稳定。由此可知,Li|LLZTO-4LiBH4|Li电池具有稳定且良好的循环性能。上述循环稳定性改善主要归因于填充的LiBH4增加了颗粒之间的表面接触,同时生成LiBO2中间层,从而避免了LiBH4和LLZTO两者之间的反应以及抑制了锂枝晶的生长[62]。 图10 60 ℃(a)和30 ℃(b)下以LLZTO-4LiBH4为电解质组装的电化学电池的恒电流循环曲线,插图为选定的循环周期内锂剥离/电镀的电压平台[62]Fig.10 Constant current cycle curves of electrochemical cells assembled with LLZTO-4LiBH4 as electrolyte at 60 ℃ (a) and 30 ℃ (b), the insets show the detailed voltage plateau of Li stripping/plating at selected cycles[62] 通过上述几种方法显著提高了硼氢化物电解质的Li+电导率。在此基础上,研究人员制备了多种硼氢化物固体电解质基全固态锂离子电池,通过电池性能测试研究硼氢化物作为固态电解质的优势和存在的问题。 Unemoto等[63, 64]以LiBH4为固体电解质,制备了高性能的固态Li-S和Li-TiS2电池,Li-S电池在45次循环后保留了64%的初始容量。Li-TiS2电池经过300个循环后容量保持率为87.8%。分析发现[65],在反应过程中,TiS2阴极与LiBH4固体电解质之间形成了Li2B12H12界面相,该界面相抗氧化稳定性优于LiBH4,可使Li+在界面上可逆、稳定地转移。 Peng等[66]用LiBH4代替液体电解质来构建稳定的固体界面,以期改善二维铋纳米片(Bi-NSs)的储锂性能。如图11所示,120 ℃时采用250 mA·g-1的电流密度循环100次后,电池仍保持较高循环容量(287 mAh·g-1),远高于采用液体电解质(~40 mAh·g-1)以及氧化物固体电解质(~180 mAh·g-1)时的循环容量。此外,Bi-NS全固态电池在开路电压下的奈奎斯特图中低频区呈现线性,而高频区没有明显的半圆形状,这表明其晶界/界面电荷呈超快速转移,界面电阻可以忽略不计,从而获得高的离子迁移速率。 图11 120 ℃、250 mA·g-1时,分别在含LiBH4电解质的Bi-NSs固态锂电池中与含LiPF6电解质的Bi-NSs液态锂电池的循环性能(a);分别以LiBH4和Li3PO4为电解质的固态锂电池:(b)循环容量比较,(c)250~4000 mA·g-1电流密度下的倍率性能,(d)开路电压下Bi-NSs液态锂电池电化学光谱的奈奎斯特图及其部分放大图[66]Fig.11 Cycling performance at 250 mA·g-1 of Bi-NSs solid lithium batteries (LIBs) with the LiBH4 electrolyte at 120 ℃, where the performance of Bi-NSs liquid LIBs with the LiPF6 electrolyte is also presented for comparison (a); solid LIBs using LiBH4 or Li3PO4 electrolytes: (b) comparison of the cycling capacity, (c) rate capabilities at various current densities from 250 to 4000 mA·g-1, (d) Nyquist plot of the electrochemical spectrum of Bi-NSs LiBs at the open circuit voltage and its partial enlargement (inset)[66] 太阳能电池的原理是借助可见光响应带隙半导体材料,将太阳能转化为便于储存和运输的电能。相对于商用晶体硅材料,有机无机杂化钙钛矿材料(AMX3,A=有机阳离子;M=Pb,Sn,Ge;X=Cl,Br,I)因光电转换效率高、易于合成且价格低廉而引起人们的关注,被认为是最具应用前景的新一代太阳能电池材料[67]。 目前,PSCs的实际应用仍受制于其内部结构稳定性。研究表明,在溶液法制备钙钛矿多晶薄膜的过程中,有机阳离子和卤化物阴离子由于其低形成能而在钙钛矿薄膜中存在大量空位点缺陷[68-70],导致器件的光伏性能差和不稳定性高。因此,如何减少离子空缺,避免有机阳离子挥发,对于提高PSCs的稳定性和效率具有重要意义。 为此,李永清课题组联合中国科学院Xu等[7]首次提出掺杂配位硼氢化物来改善PSCs的光电转换性能和稳定性。如图12所示,MAPbI3(MA=CH3NH3+)中固有的碘离子空位被超卤素基团(BH4-)部分取代,形成MAPbI3-x(BH4-)x钙钛矿结构。这不仅弥补了碘离子空位,还借助双氢键作用抑制了甲胺基的分解和流失,从而获得光滑且致密的低缺陷薄膜。结果表明,与初始MAPbI3相比,所制备的MAPbI3-x(BH4-)x具有较好的综合性能:光电转换效率(PCE)从18.43%显著提高到21.10%,短路电流密度(Jsc)从21.71提高到23.89 mA·cm-2,开路电压(Voc)从1.08提高到1.11 V,此外,还显著提高了钙钛矿薄膜的热稳定性和紫外光照稳定性。 图12 薄膜太阳能电池器件结构示意图(a);BH4-的分子结构(b);超级卤素BH4-组装在碘离子空位上,与甲基铵(CH3NH3+)形成氢键(c)[7]Fig.12 Schematic architecture of the thin film solar cells device (a); molecular structure of BH4-(b); super-halogen BH4- assembled on the iodide ion vacancy, forming hydrogen bonding with methylammonium (CH3NH3+) (c)[7] 基于配位硼氢化物材料自身的物理化学特性,人们对其在能量储存和转换领域展开了多方面应用研究,包含固态储氢、水解制氢、固态电解质和太阳能电池,所取得的科学进展令人印象深刻。同时,不可否认,配位硼氢化物的多功能应用仍然存在诸多挑战。 为了实现安全、高效的能源转换与存储的目标,推进硼氢化物实用化进程,亟需深入开展以下几方面工作:① 对硼氢化物材料的基本物理化学特性有待进一步理解和掌握,尤其是借助中子衍射、同步辐射和强磁场等国家大科学装置对其原子占位、电子分布、离子迁移等进行原位观察和解析;② 调控硼及其伴生物的惰性问题,从根本上来解决固态储氢和水解制氢的可逆再加氢问题;③ 借助机器学习来理论指导硼氢化物的晶体结构设计和调控,构建离子快速迁移的低能通道,以提升全固态电池和太阳能电池的转换效率和稳定性;④ 继续拓展和丰富硼氢化物的新功能性应用,并揭示和完善其内在作用机制。上述问题的解决将极大地推动功能性硼氢化物应用,实现电能和氢能的高效存储与转换,同时为探索硼氢化物作为其他功能性材料提供新启示。
5 太阳能电池
6 结 语
