取代环己酮还原立体选择性的过渡态理论解释
2022-05-12李佳胡潇王健春于凯歌耿诗宁孟祥福
李佳,胡潇,王健春,于凯歌,耿诗宁,孟祥福
首都师范大学化学系,北京 100048
取代环己酮的还原具有立体选择性,但仅从动力学和热力学角度(即“产物发展控制”,product development control)去预测取代环己酮还原产物是不准确的,一直以来没有广泛地被人们接受,而“立体途径控制”(steric approach control)却有很好的理论解释,可以归结为“NaBH4或LiAlH4还原环己酮,如果羰基不受阻碍,生成赤道醇产物;如果受到阻碍或阻碍非常强烈时,生成轴向醇产物”,这是巴顿关于取代环己酮还原产物预测的总结[1],在今天仍能得到验证并很好地使用。但是,这仅是从反应物或者产物的角度考虑。早在1970年,Eliel等人就发现环己酮还原过程中形成的过渡态对产物具有一定的影响并以此作为预测产物的依据[2]。后来,Andrzej和Neufeldt等人通过还原过渡态的电子效应以及过渡态构象很好地解释并预测了环己酮还原产物的立体选择性[3,4]。研究表明,利用取代环己酮还原过程中形成的过渡态空间位阻来预测还原产物立体选择性会更准确,同时过渡态的电子效应和优势构象也可以作为有利依据分析过渡态的存在形式,进而判断形成产物的立体构型[5-7]。
大量文献表明,虽然对NaBH4还原环己酮的机理还未形成更为完善的体系,但是由于对LiAIH4和其他试剂的还原机理还不清晰,所以下面引例主要选取已经证实的NaBH4的还原机理进行解释,部分引例选取LiAlH4简单讨论。
1 空间效应
环己酮还原过程中,空间效应表现为还原试剂和反应底物的位阻大小。在考虑取代环己酮的空间位阻时,对还原试剂1,2-二轴相互作用和1,3-二轴相互作用的考虑必不可少。以NaBH4为例,除了H-进攻外,同时参与反应的还有剩余的硼氢化物分子(图1a,b)。当H-沿赤道向进攻时,不会受到C-2和C-6直立氢的影响,但是过渡态中待形成的-OH会与3号位的轴向氢存在相互作用,而沿轴向进攻时会存在C-2和C-6两个轴向氢对还原剂的阻碍作用。相反,如果C-3有取代基,则进一步考虑3号位取代基对还原剂的作用。如图1c,d所示,3,3,5-三甲基环己酮的C-3直立键的甲基会阻碍还原剂的轴向进攻,产物的立体选择性需要综合考虑赤道向和轴向受阻碍程度的相对大小来预测。如表1所示,当位阻较小的还原剂(如NaBH4,LiAlH4)进攻取代环己酮时,还原剂主要从轴向进攻。例如,4-叔丁基环己酮被NaBH4和LiAlH4还原时,赤道醇的产率分别为80%和92%,此时1,3-二轴相互作用不明显。随着还原剂的体积增大以及羰基的空间位阻效应增强,还原剂主要从赤道方向进攻。例如,2-甲基环己酮被LiAl(OtBu)3H还原成赤道醇的产率为64%,当空间位阻进一步增加,3,3,5-三甲基环己酮还原成赤道醇的产率仅为5%,这说明赤道向进攻过渡态中1,3-二轴存在较大的空间位阻效应,对环己酮还原产物的立体选择性起到决定作用。

表1 不同还原剂(亲核试剂)和反应底物的赤道醇的比例[9–11]
2 电子效应
由于羰基碳氧双键p轨道两侧电子密度是不对称的,赤道方向电子密度更为紧密,轴向电子密度较为疏松[8],所以还原剂倾向于从轴向(电子密度小的一面)进攻,形成更好的轨道重叠,因此电子效应对取代环己酮还原立体选择性有重要作用。
取代环己酮被还原剂进攻时的电子效应主要表现为还原剂进攻羰基时生成初生键[3]。σ轨道与邻近最低空σ*轨道形成二电子稳定相互作用以及初生键的σ*轨道与邻近最高占有σ轨道形成二电子稳定相互作用(如图2所示)。
2.1 初生键的σ轨道与邻近最低空σ*轨道形成二电子稳定相互作用
如图3a所示,以4-叔丁基环己酮为例,当叔丁基位于直立键时,在被LiAlH4还原时得到的几乎全部为轴向进攻产物。
这是因为当还原剂沿轴向进攻4-叔丁基环己酮的时候,形成的初生键σ轨道与C4直立键取代基C―C反键轨道发生较大重叠(图3b),从而使电子离域,满足二电子稳定效应[3],稳定了过渡态。而当还原剂沿赤道向进攻4-叔丁基环己酮的时候(图3c),无法形成二电子离域。

图3 (a)LiAlH4还原4-叔丁基环己酮;(b)还原剂沿轴向进攻过渡态;(c)还原剂沿赤道向进攻过渡态
但是,叔丁基位于平伏键是其优势构象,所以我们着重介绍该构象的还原立体选择性情况。相较而言,稳定构象的4-叔丁基环己酮在被格氏试剂加成时得到的大多是轴向产物,如表2所示。这是因为烷基负离子从轴向进攻4-叔丁基环己酮时形成的初生键σ轨道与C4平伏键取代基C―C σ*轨道无法实现较大重叠,使电子离域,达到二电子稳定状态。而当烷基负离子从赤道向进攻时形成的初生键σ轨道与C4平伏键取代基C―C σ*轨道可以形成较大的重叠。由此可见,亲核试剂进攻方向需要满足初生键的σ轨道与邻近最低空σ*轨道形成二电子稳定相互作用,稳定过渡态。
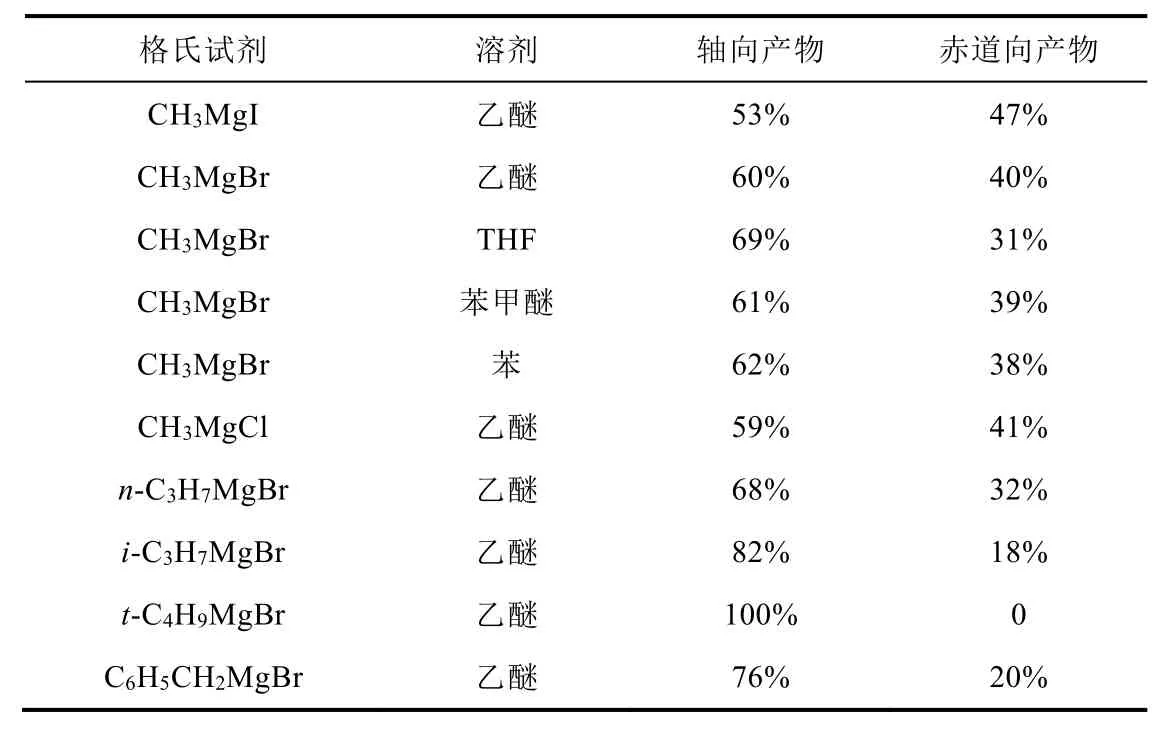
表2 烷基格氏试剂与4-叔丁基环己酮的加成反应[5]
2.2 初生键的σ*轨道与邻近最低空σ轨道形成二电子稳定相互作用
当还原剂沿轴向进攻稳定构象的4-叔丁基环己酮时,形成的初生键σ*轨道和C4平伏键取代基C―C的σ成键轨道发生重叠(图4a),达到二电子稳定状态,稳定过渡态。而当还原剂沿赤道方向进攻稳定构象的4-叔丁基环己酮时,虽然形成的初生键σ*轨道也会和C4取代基的C―C成键轨道发生重叠(图4b),但重叠程度较轴向进攻时小,二电子离域效应较差。
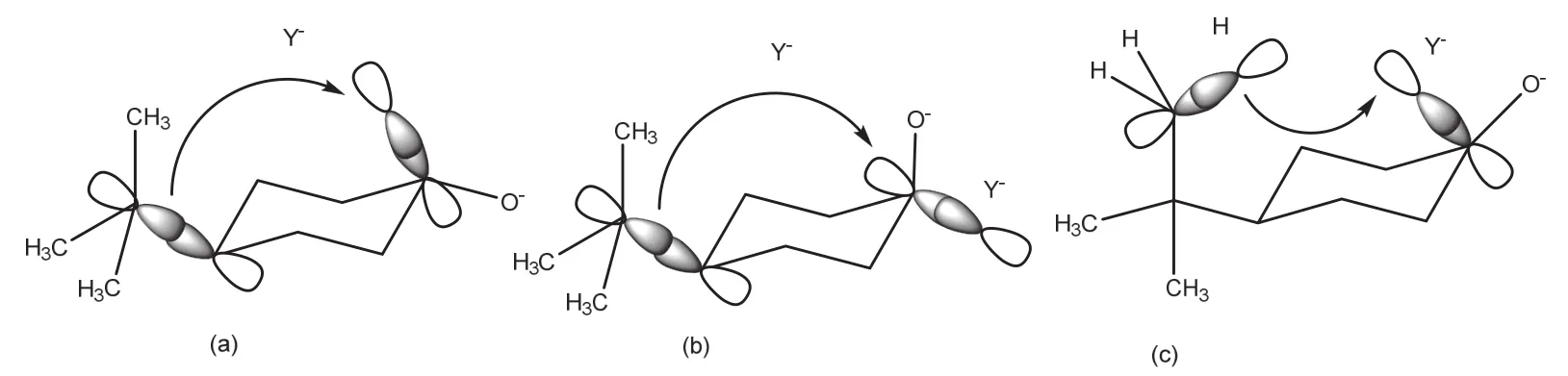
图4 (a)轴向进攻过渡态:C―C σ成键轨道与初生键的σ*轨道的重叠;(b)赤道向进攻过渡态:C―C σ成键轨道与初生键的σ*轨道的重叠;(c)轴向进攻过渡态:C―H键的成键轨道与初生键的σ*轨道
此外,由于叔丁基的空间延展较大,故存在叔丁基支链甲基的C―H键的成键轨道与初生键的σ*轨道发生进一步重叠(图4c)。这在很大程度上将C―H键的电子反馈到初生键的反键轨道上,使二电子离域相互作用更大。因此,反应更倾向于沿轴向进行。
3 构象
如果仅考虑电子效应和空间效应不能充分解释取代环己酮还原的立体选择性,还需要从结构的角度来进行考虑,即构象分析。下面,我们从扭转张力和构象稳定性两个方面进行讨论,使用到的工具是:Felkin-Anh模型(图5a)和标准交叉构象偏转角(图5b)平均值及公式。

图5 (a)Felkin-Anh模型;(b)标准交叉构象
3.1 扭转张力
Feklin-Anh规则是:RL与羰基处于垂直交叉式;亲核试剂从空间位阻较小一侧,并与羰基呈107°的方向进攻。在这里我们考虑环己酮C-2和C-6处轴向取代情况,还原剂在接近羰基时会与取代基重叠,产生扭转应变,所以在形成过渡态时可以通过键的旋转使扭转张力减小。
以NaBH4为例(图6),在赤道向进攻时形成的C―H键与C-2、C-6轴向氢产生位阻影响。而在轴向进攻环己酮时,扭转应变会起到更重要的作用,这是因为羰基被C-2和C-6轴向氢重叠,当沿轴向进攻时,扭转张力会缓解;当沿赤道方向进攻时,为了使氧原子形成完全重叠的排列会增加扭转张力[12]。

图6 轴向进攻和赤道向进攻的纽曼投影式[12]
从椅式构象出发,尽管赤道向进攻和轴向进攻形成的过渡态相似,但是赤道向进攻形成C-H键更加滞后。如图7a,b所示,轴向进攻使C―O键与垂直方向的角度更小,形成交错式构象(图7c,d),没有环张力;赤道向进攻使角度更大,不仅会使O―C―C―C二面角增大,而且为了达到交错式构象而增大环张力。

图7 (a)轴向进攻;(b)赤道向进攻;(c)轴向进攻C2C1方向纽曼投影式;(d)赤道向进攻C2C1方向纽曼投影式[13]
3.2 构象稳定性
当LiAlH4还原稳定构象的4-叔丁基环己酮时,主要产物为轴向进攻产物,还原过程中过渡态以六元环形式存在(图8)。而对于过渡态构象a和b,可以通过对标准交叉构象的偏转角的平均值来讨论其稳定性。Neufeldt等人[4]通过式(1)计算出平均偏转角θa=12°,θb=14°:

可见,图8b构象的平均偏转角较小,扭转角度小,过渡态较稳定。同时,也可以从能量角度加以证明,a构象的能量比初始反应物高72.7 kJ·mol-1,b构象能量高77.3 kJ·mol-1[4],这说明LiAlH4还原稳定构象环己酮时主要产物为轴向进攻产物。

图8 LiAlH4还原4-叔丁基环己酮形成的过渡态
当LiBH(i-Pr)3还原稳定构象的4-叔丁基环己酮,主要产物为赤道向进攻产物,如图9所示。

图9 LiHB(i-Pr)3还原4-叔丁基环己酮
与上述不同,LiBH(i-Pr)3的还原过程经历五元环过渡态(图10)。通过对两种构象偏转角分析,发现θ赤道向=10°,θ轴向=11°,可以看出轴向过渡态的偏转角较赤道向略大。但是通过能量数据比较,赤道向进攻形成的过渡态能量比初始反应物高出103.2 kJ·mol-1,而轴向时的能量却高出118.7 kJ·mol-1[4]。由于能量对于轴向过渡态影响较大,造成了构象不稳定,所以LiBH(i-Pr)3还原主要得到赤道向进攻产物。

图10 LiBH(i-Pr)3还原4-叔丁基环己酮形成的过渡态
4 结语
通过上述讨论得知,从过渡态稳定性的角度去解释取代环己酮还原立体选择性是有利的。在取代环己酮还原反应过程中,不仅存在动力学和热力学控制因素,更重要的是过渡态空间位阻和电子效应与构象稳定性的竞争。在空间位阻效应中,未阻碍羰基的取代环己酮主要受到还原剂轴向进攻,而受阻取代环己酮主要受到还原剂赤道向进攻。此外,在过渡态中存在着二电子稳定相互作用,通过初生键σ成键轨道和σ*反键轨道分别与n电子以及饱和σ键和π键的距离体现,这一点在预测反应产物的类型上也有很重要的作用。在过渡态构象上,通过扭转张力和键的夹角理论计算,可以分别分析其过渡态扭转张力的相对大小和相对于标准构象的偏转程度,同时通过能量数据对比,也可以判断出产物的主要存在形式。总之,从过渡态空间效应、电子效应和构象这三种因素出发,可以很好地解释并预测不同金属氢化物还原取代六元环酮产物的立体选择性,这对有机合成有很大的帮助,可以通过实验前的预测,节约时间,提高合成效率。
