灰化- 酸分解碘量法测定含树脂铜原料中铜含量
2022-05-12田强坤施艳艳杨琳娟左仕新
田强坤, 李 超, 施艳艳, 李 娟, 杨琳娟, 左仕新
(楚雄滇中有色金属有限责任公司, 云南 楚雄 675000)
云南某冶炼厂进厂铜原料中有部分批次原料中含树脂粉末,对该批次原料制成的成分检测样品铜含量进行测定,在检测前处理过程中,发现样品分解不完全,分析结果重复性、准确性也均不符合评定要求。对该类样品进行电镜扫描,发现树脂粉末中夹杂有单质铜。
目前,含铜原料中常用的铜含量测定方法较多,除国家标准方法碘量法[1]和电解法[2]外,还有黎香荣等[3]提出的光度滴定法、杨丽飞等[4]提出的高压密封微波消解法及其他一些酸分解试样处理方法[5-10]。廉惠萍[11]提出的酸处理阶段加入氟化氢铵对含硅较高试样、硅铝晶体包裹试样的处理方式,有效解决了难溶元素硅对样品中铜含量测定的影响。现有方法虽测量原理不尽相同,但均以将试样中待测元素铜的不同形态转化为Cu2+为测定基础。然而,对于含树脂的铜原料,因树脂具有良好的耐酸碱性质,几乎不与所有的酸碱溶剂发生反应,从而隔绝了树脂粉末中夹杂的单质铜与样品分解试剂接触的机会,故采用上述测定方法均不能将试样分解完全,存在分析结果重复性、准确度无法满足要求的问题。因此,如何使试样分解完全是含树脂铜原料中铜含量准确测定的关键。
本文采用灰化- 酸分解法对试样进行处理,实现了含树脂铜原料样品的完全分解,再衔接碘量法进行滴定,取得了精密度好、准确度高的分析结果,且该处理方式成本低、操作简单,具有推广应用价值。
1 试验设备及试剂
1.1 分析试样
1) 铜精矿标准物质GBW07166,来源于地球物理地球化学勘查研究所,ω(Cu)=24.20%。
2) 含树脂铜原料,来源于配混矿,主要成分:Cu 12%~16%,Fe 18%~20%,SiO211%~14%,CaO 4%~7%,Al2O32%~4%,树脂8%~12%。
3) 大矿山铜精矿,来源于使用浮选工艺生产的铜精矿,主要成分:Cu 10%~32%,Fe 20%~35%,SiO21%~20%,CaO 1%~5%,Al2O31%~5%。
1.2 主要仪器
试验用到的主要设备为5E- MF6100K 智能马弗炉(开元仪器),最高使用温度999 ℃, 温度分辨率 0.1 ℃,可自由设置升温时间。
1.3 主要试剂
1)氢氟酸(AR),浓度为1.15 g/mL。
2)盐酸(AR),浓度为1.19 g/mL。
3)硝酸(AR),浓度为1.42 g/mL。
4)高氯酸(AR),浓度为1.67 g/mL。
5)溴(AR)。
6)硝硫混酸。由700 mL硝酸(AR)和300 mL硫酸(AR,浓度1.84 g/mL)混合配制而成。
7)乙酸- 乙酸铵缓冲溶液(300 g/L)。称取90 g乙酸铵(AR),置于400 mL烧杯中,加入150 mL水和100 mL冰乙酸(AR,浓度1.05 g/mL),溶解后,用水稀释至300 mL,混匀,溶液pH值为5。
8)氟化氢铵饱和溶液,贮存于聚乙烯瓶中。
9)碘化钾(AR)。
10)淀粉(AR)溶液,浓度为5 g/L。
11)三氯化铁(AR)溶液,浓度为150 g/L。
12)硫氰酸钾(AR)溶液(100 g/L)。称取10 g硫氰酸钾于400 mL烧杯中,加入100 mL水溶解,再加入2 g碘化钾溶解,然后加入2 mL淀粉溶液,并滴加碘溶液(约0.04 mol/L)至恰好呈蓝色,再用硫代硫酸钠标准滴定溶液滴定至蓝色刚好消失。
13)高纯铜片,ω(Cu)≥99.999%,使用前进行前处理,方法为将铜片置于微沸的乙酸中,微沸1 min,取出后用水和无水乙醇分别冲洗两次以上,在100 ℃烘箱中烘4 min,冷却,置于磨口瓶中备用。
1.4 硫代硫酸钠(AR)标准滴定溶液的配制和标定
1.4.1 配制
称取100 g五水硫代硫酸钠(Na2S2O3·5H2O)置于加有500 mL无水碳酸钠(AR)(4 g/L)的1 L烧杯中,溶解后移入10 L棕色试剂瓶中;用煮沸并冷却的蒸馏水稀释至约10 L,加入10 mL三氯甲烷(AR),静置2周;使用时过滤,补加1 mL三氯甲烷,混匀,静置2 h,标定。该溶液每隔1周必须重新标定一次。
1.4.2 标定
1)方法一。称取3份经前处理的高纯铜片0.05~0.06 g(精确至0.000 01 g)。分别置于250 mL锥形瓶中,加入15 mL硝酸(1+1)(1+1指的是1体积硝酸和1体积水的混合溶液),低温分解完全,溶至体积约1 mL,取下,稍冷,吹水约30 mL,加入0.5 g尿素,煮沸取下至室温。加入1 mL三氯化铁溶液(150 g/L)混匀,滴加乙酸- 乙酸铵溶液(300 g/L)至红色不再加深并过量3~4 mL,然后滴加氟化氢铵溶液(250 g/L)至红色消失并过量2 mL,摇匀,沿杯壁吹少量水。向溶液中加入2 g碘化钾,轻摇使其混匀,立即用硫代硫酸钠标准滴定溶液滴至浅黄色,加入2 mL淀粉溶液(5 g/L),继续滴定至浅蓝色,加入2 mL硫氰酸钾溶液(400 g/L)激烈振摇至蓝色加深,再滴定至蓝色恰好消失为终点。随同做空白试验,以消除系统误差。
2)方法二。移取3份25.00 mL浓度2.0 mg/mL铜标准贮存溶液分别置于250 mL锥形瓶中,各加入5 mL硝酸,于电热板低温处蒸至溶液体积约1 mL。取下稍冷,用约30 mL水吹洗杯壁,冷至室温后进行标定。
平行标定三份,其极差值不大于5×10-5mol/L时,取其平均值,否则重新标定。
2 试验原理及方法
2.1 试验原理
试样按升温程序经过200 ℃固化、500 ℃碳化、900 ℃灰化后,可以破除试样中树脂,使其以CO2气体形式溢出,从而使包裹的铜暴露;再经盐酸、硝硫混酸分解;然后用乙酸铵溶液调节溶液pH值为3.0~4.0,用氟化氢铵掩蔽铁,加入碘化钾与二价铜作用,析出的碘以淀粉为指示剂,用硫代硫酸钠标准滴定溶液滴定。
2.2 试验方法
试验方法中所指试样为经过研磨且粒度不大于100 μm的样品。
2.2.1 固化- 碳化- 灰化
称取0.2 g试样,精确至0.000 1 g,小心转移至垫有6层Φ30 mm定性滤纸的30 mL瓷坩埚中,并在样品上方覆盖6层Φ20 mm定性滤纸,压实,采用马弗炉固化- 碳化- 灰化样品,具体程序见表1。

表1 马弗炉固化- 碳化- 灰化样品的温度控制程序
2.2.2 酸分解
灰化结束后,取出坩埚,冷却后将样品转移到250 mL烧杯中,用水润湿,加入10 mL盐酸;在瓷坩埚中滴入1~2滴HF,5 mL盐酸,低温溶解至1 mL左右后加入5 mL硝酸,溶解并蒸发至体积2 mL左右后,将溶液转入对应盛装试样的250 mL烧杯中,并用5 mL硝酸冲洗坩埚内壁及杯口两次并转移至对应250 mL烧杯中。将烧杯置于低温电炉上加热3~5 min,取下稍冷,再加入15 mL硝硫混酸和1 mL溴,加热至冒白烟时取下冷却,加入5 mL高氯酸,溶解并蒸发至近干。
需要注意的是,若试样中含硅较高时,需在低温加热前加入0.5 g氟化氢铵。
2.2.3 滴定
用30 mL水吹洗表皿和杯壁,盖上表皿,置于电热板上煮沸,取下冷至室温,滴加乙酸- 乙酸铵溶液至红色不再加深并过量3~5 mL;然后滴加氟化氢铵饱和溶液至红色消失并过量1 mL,混匀;加入2~3 g碘化钾,摇动溶解,立即用硫代硫酸钠标准滴定溶液滴定至浅黄色,加入2 mL淀粉溶液,继续滴定至浅蓝色;加入5 mL硫氰酸钾溶液,剧烈摇振至蓝色加深,再滴定至蓝色刚好消失计为终点。随同试样进行空白试验。
2.2.4 计算
铜质量分数计算式见式(1)。
(1)
式中:ω(Cu)为铜质量分数,%;c为硫代硫酸钠标准滴定溶液浓度,mol/L;V1为试料消耗硫代硫酸钠标准溶液的体积,mL;V2为空白溶液消耗硫代硫酸钠标准滴定溶液的体积,mL;m0为试料质量,g;63.55为铜摩尔质量,g/mol。
3 结果与讨论
3.1 灰化温度- 时间的影响
为确定灰化温度- 时间对样品分解及分析结果的影响,试验选用3个含树脂样品(TJK- 01,TJK- 02,TJK- 03),分别采用3种方式进行试样处理。
1)无灰化过程。试料滴加1~2滴HF,15 mL盐酸,低温溶解至体积3~5 mL,取下稍冷,加入15 mL硝硫混酸和1 mL溴,加热至冒白烟时取下冷却,加入5 mL高氯酸,溶解至近干。
2)快速灰化。样品小心转移至垫有6层滤纸的30 mL瓷坩埚中,并在样品上方覆盖6层滤纸,采用表2温度- 时间程序进行灰化,灰化结束后按2.2.2~2.2.4步进行。
3)缓慢灰化。按2.2.1~2.2.4步骤对样品进行处理。各试样分别平行测定5次,试验结果见表3。

表2 快速灰化温度- 时间程序

表3 不同灰化温度- 时间程序下分析结果
表3数据表明,不灰化的样品分析结果较快速灰化及缓慢灰化严重偏低,其测定结果的相对标准偏差(RSD,n=5)高达10.02%~11.96%,精密度差,原因是未经灰化样品中的树脂大分子有机聚合物包裹的铜未全部反应转化为 Cu2+;快速灰化的样品测定结果较不灰化有所提高,但测定结果的相对标准偏差(RSD,n=5)为2.11%~3.80%,精密度仍不理想,原因一方面是升温过快,样品、垫层滤纸和覆盖滤纸迅速爆燃,致使样品损失,另一方面是因为灰化时间短,灰化不完全,仍有部分树脂大分子有机聚合物存在,致使后续酸溶过程样品分解不完全;缓慢灰化的样品,分析结果重复性好,精密度高。
缓慢灰化的样品分析结果精度高的机理为:样品在200 ℃下固化,避免后序碳化、灰化过程中造成粉末损失;样品在500 ℃下碳化,在缺氧状态下将树脂大分子有机聚合物全部碳化为碳质;样品在900 ℃下灰化,在有氧状态下使碳质全部灼烧生成二氧化碳逸出,完全消除树脂有机物对铜溶解的影响。因此,在对含树脂铜原料中的铜含量进行检测分析时,需先对样品进行固化- 碳化- 灰化处理,且过程中应严格控制温度和时间,以避免灰化不完全和样品喷溅损失。
3.2 滤纸层的选择
为确定滤纸垫层及覆盖层的最佳数量,试验选用含树脂样品TJK- 01,分别垫入及覆盖0层、2层、4层、6层、8层、10层滤纸,按表1程序进行灰化处理,按照上文试验方法步骤进行处理及测定,分别平行测定5次,滤纸垫层及覆盖层数量对测定结果的影响见表4。
由表4可知,当滤纸垫层及覆盖层数量为0层、2层、4层时,灰化完成后样品与坩埚底部有不同程度的粘接,铜测定结果偏低,且精密度较差,这是因为滤纸垫层及覆盖层数量过少,在灰化过程中样品与坩埚底部粘结,转移不完全所致。当滤纸垫层及覆盖层为6层、8层、10层时,灰化完成后样品与坩埚底部基本无粘接,铜测定结果趋于平稳,且多次分析结果精密度好。但加入滤纸垫层及覆盖层数量较多时,灰化后灰分较多,增加酸分解难度,故确定滤纸垫层及覆盖层的数量分别为6层。

表4 滤纸垫层及覆盖层数量对测定结果的影响
4 样品分析
4.1 方法的精密度试验
试验选用6个试样(3个为含树脂铜原料,3个为大矿山铜精矿)及1个国家一级标准物质GBW07166,按照试验方法进行测定。每个样品平行测定5次,验证本方法的精密度和准确度。试验结果见表5。

表5 方法的精密度和准确度试验结果
4.2 加标回收试验
试验选用上述精密度试验的3个为含树脂铜原料作为本底样品,以国家一级标准物质GBW07166作为加标样品,进行样品加标回收试验,通过回收率,再次验证方法准确度,结果见表6。
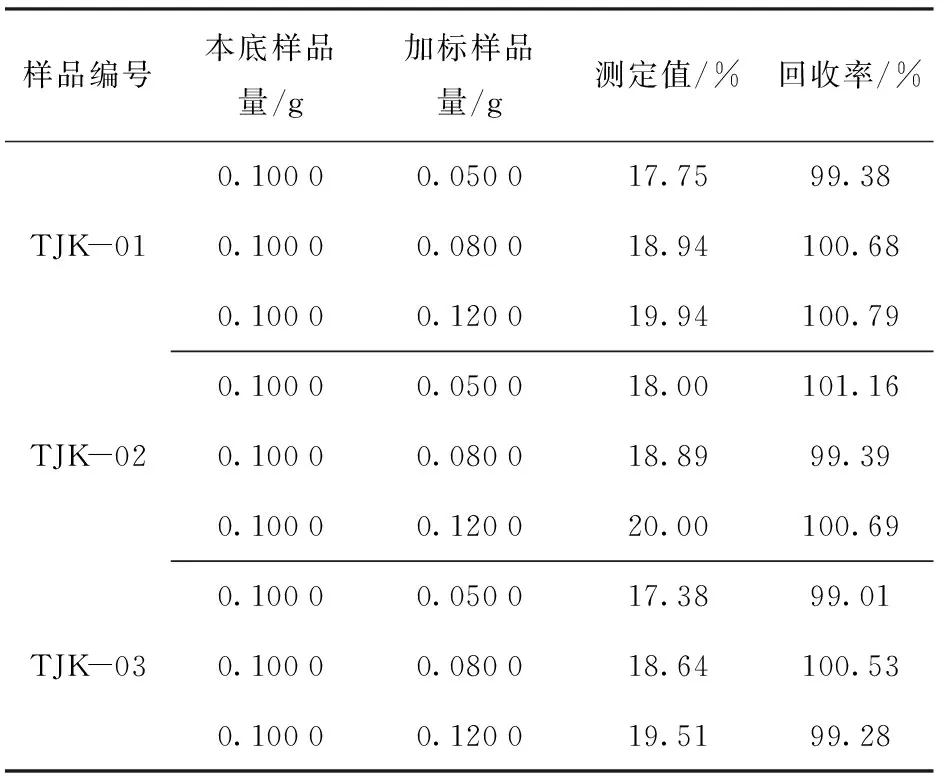
表6 加标回收试验结果
由表5、表6可以看出:本方法测定结果的相对标准偏差(RSD,n=5)为0.20%~0.41%,加标回收率为99.01%~101.16%,相对误差RE为0.06%~0.27%,精密度好、准确度高,满足分析检测要求。
5 结论
针对目前检测方法在含树脂铜原料中铜含量测定中存在的问题,本文在总结分析国家标准方法和其他一些湿法处理方式的基础上,创新性地采用灰化- 酸分解的方法对被检测样品进行处理,得到了满意的测定结果。
1)灰化- 酸分解方法包括灰化和酸解两部分。灰化即于200 ℃固化、500 ℃碳化、900 ℃灰化,目的是使样品中树脂结构完全破坏,以消除对铜溶解的影响;酸解是采用强酸对样品进行分解,尽量使得铜溶解完全。
2)在灰化过程中,对滤纸垫层及覆盖层进行了试验,结果表明,滤纸垫层和覆盖层分别为6层比较合适。
3)在试验条件精准控制的前提下,对样品进行了精密度、准确度及加标回收试验,结果表明,本试验方法测定结果的相对标准偏差(RSD,n=5)为0.20%~0.41%,加标回收率为99.01%~101.16%、相对误差RE为0.06%~0.27%,精密度和准确度高,满足分析检测要求。
