基于外电场作用氟氯碳酰的光谱特性和解离特性
2022-05-12闫好奎布玛丽亚阿布力米提郑敬严
向 梅, 安 桓, 闫好奎, 布玛丽亚·阿布力米提, 郑敬严
(1. 新疆师范大学 物理与电子工程学院, 乌鲁木齐 830054; 2. 新疆计量测试研究院, 乌鲁木齐 830011)
1 引 言
1974年Molina团队在Nature期刊上发表关于氟利昂对臭氧层有很大的破坏性[1],1985年经Joe Farman团队通过在南极站观测进一步证实此结论后[2],人们对臭氧层的保护越来越重视. 科学家研究发现不仅氟利昂会破坏臭氧层[3],哈龙分子等含卤素分子解离后也会破坏臭氧层[4, 5]. 在加外电场作用下,含卤素分子会发生一系列物理和化学变化[6-8],例如分子化学键断裂,分子轨道、晶体感应、激发光谱等发生变化[9-11]. 因此目前利用加外电场来研究分子的物理性质成为当下理论研究的热门领域之一[12-16].
三体解离动力学因可提供解离过程的动态演化信息而受到研究者持续广泛的关注[17-20]. 从化学的角度来看,多原子分子或离子的三体解离是分子中两个特定化学键断裂的过程,其中断键时序至关重要,它决定着解离过程中三体解离碎片间相互作用的关联程度. 根据时间差(τ)和待解离中间体碎片的转动周期(Tr)的相对关系,可将三体解离区分为顺序解离(stepwise dissociation)和协同解离(concerted dissociation);根据τ/Tr和1的大小关系又可以将解离细分为顺序解离(τ/Tr>1)、同步解离(τ/Tr=0)和非同步解离(0<τ/Tr<1). 目前研究分子的协同解离现象主要通过实验手段,Pragya团队采用离子分子碰撞装置研究了OCS3+的三体解离,结果显示能量为200kV/q的Xe9+离子与OCS分子多次碰撞使分子发生协同解离和逐步解离[21];刘玉柱团队通过飞行时间质谱技术研究了氟利昂1110在800 nm飞秒脉冲光作用下的多光子解离,采用离子速度成像技术应证了C2Cl3+与 C2Cl2+分别对应逐步解离和协同解离通道[22];Shin-Huang团队观察到丙烯醛在193 nm的同步辐射光源激发下,探测到协同解离碎片C2H2+CO+H2,并利用量子化学计算确定了基态势能面上的相应跃迁结构,该研究为同步协同三体解离提供了新的机制,并将光化学研究扩展到通过打破非等价化学键的同步协同多重解离[23];刘博通团队通过直流(dc)切片速度映射成像技术研究了二氟二溴甲烷分子与800/400 nm飞秒激光场之间的相互作用,通过分析碎片离子的动能释放分布和角度分布,在400 nm激光场中确定了两个C-Br键的异步断裂现象[24]. 采用理论计算的方法也可以研究分子的协同解离现象,Houk团队在密度泛函理论B3LYP/6-31G(d)基组水平上采用Cope重排研究了1,5-己二烯的解离现象,发现了95%的协调解离和5%逐步解离轨迹[25];刘玉柱团队借助Gaussian计算软件通过密度泛函理论B3PW91/6-311+G (2df)基组计算了二氟二溴甲烷的势能面,研究发现外电场强度为0.055 a.u.时两个C-Br键依次解离,外电场强度为0.065 a.u.时协同解离现象发生[26].
氟氯碳酰作为生产聚氨酯的中间体和合成药物的试剂被广泛使用于化学工业领域. 由于氟氯碳酰分子在云层中会水解出氯元素,与大气中的臭氧层发生连锁反应(Cl+O3→ClO+O, ClO+O3→Cl+2O2)[4],造成对臭氧层的破坏. 在平流层中已检测到光气(COCl2)、氟氯碳酰(COFCl)和碳酰氟(COF2)[27],对流层中也检测到COCl2和COF2,并且观测其浓度显著增加[27]. 目前,Heineking团队研究了COFCl四种同位素异构体的转动光谱,确定了它们的振动-旋转相互作用常数、它们各自在喷流中的振动温度,评估了四极耦合张量的主值及其在键轴系统中的位置[28];Christof团队通过共振增强多光子电离和飞秒时间技术在解离波长235 nm附近监测氯碎片,来研究氟氯碳酰的解离现象,首次通过实验测定了COFCl生成热和C-Cl键断裂的离解能为-397±15 kJ/mol和364±8 kJ/mol,但并没有观察到协同解离现象[29];Karl-Heinz团队通过共振增强多光子电离结合飞行时间测量(REMPI-TOF)研究了COFCl的解离现象,并观察到快速的异步协同解离和缓慢的同步协同解离[30]. 由此可见,目前对COFCl分子是否发生协同解离仍存在争议. 本文通过密度泛函理论(Density functional method,DFT)在B3LYP/6-31G(d)水平上优化计算了不同外电场(-0.02—0.07 a.u.)作用下氟氯碳酰分子的基态几何结构、能隙、红外光谱、拉曼光谱以及解离特性,尝试观察其协同解离现象,以期为COFCl分子在外电场降解提供必要的理论依据.
2 理论及计算方法
在外电场作用下氟氯碳酰分子体系哈密顿量H为:
H=H0+Hint
(1)
其中H0为无外场时的哈密顿量,Hint为外电场与分子体系的相互作用的哈密顿量. 在偶极近似下,分子体系与外电场F的相互作用能为:
Hint=-μF
(2)
其中μ为分子的电偶极矩,F代表点电荷模型或者有限场模型下的电场强度,其中1a.u.=5.14225×1011V/m. 收敛条件分为SCF收敛和Davidson迭代收敛:其中SCF收敛为Maximum Force≤0.00045,RMS Force≤0.0003, Maximum Displacement≤0.0018, RMS Displacement≤0.001200;Davidson迭代收敛为所要计算的n个态的波函数变化量小于0.001.
本文通过不同的方法和基组模拟多种情形,对氟氯碳酰分子进行结构优化计算,将计算得到的键长、键角与实验值作比较,选取出最合适的DFT/B3LYP方法6-31G(d)基组;通过外加不同电场(-0.02—0.07 a.u.)条件下,计算分析氟氯碳酰分子的几何构型、偶极矩、电荷分布、振动频率、轨道能级分布、红外光谱、解离势能面的变化情况. 本文的理论计算通过Gaussian 09[31]软件完成.
3 结果与讨论
3.1 氟氯碳酰分子的基态稳定构型
在无外电场作用下,本文对氟氯碳酰分子不同方法和基组进行优化计算,将优化出的平衡核间距R、键角A与实验值做比较,结果如表1所示. 表中数据表明计算密度泛函理论B3LYP/6-31G(d)基组得到的平衡核间距、键角与实验数据基本相符,因此,沿着箭头所示方向加外电场时均选用DFT/B3LYP方法6-31G(d)基组进行模拟计算. 建立如图1所示的建立笛卡尔坐标系.
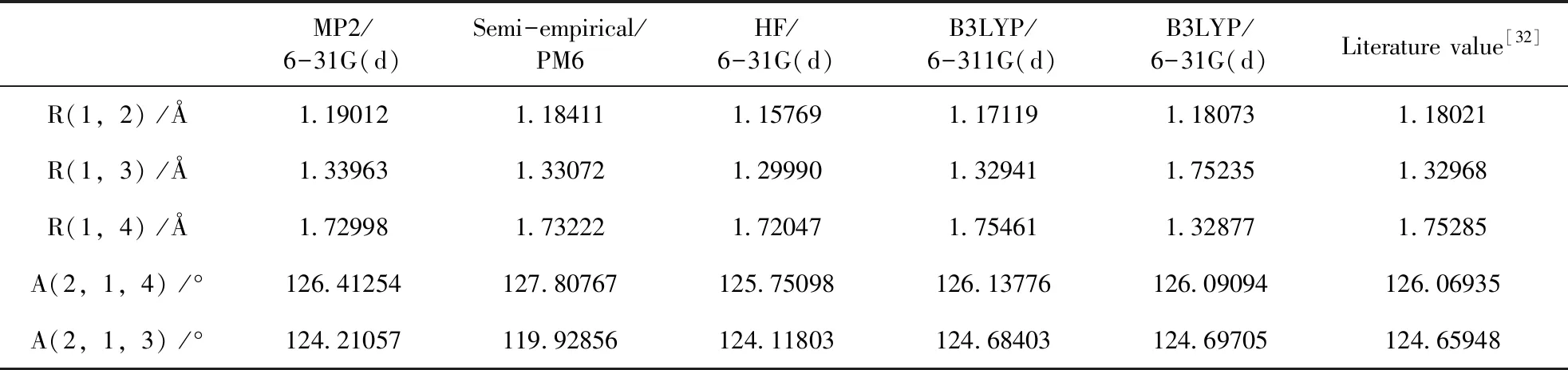
表1 不同方法优化氟氯碳酰分子基态结构
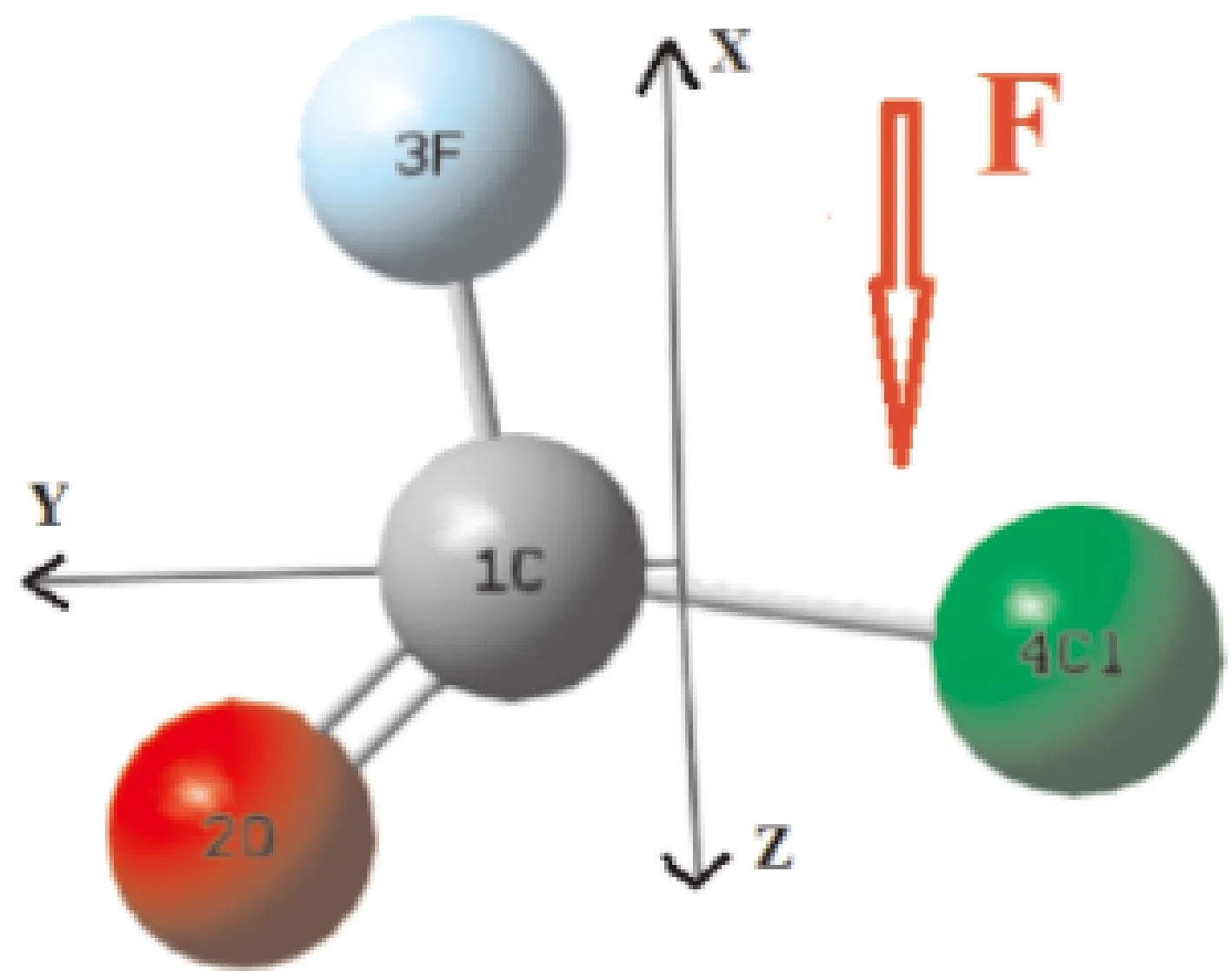
图1 无外电场下的氟氯碳酰分子稳定基态构型Fig. 1 The stable ground state configuration of carbonyl chloride fluoride molecule without external electric field
3.2 外加电场下氟氯碳酰分子的键长和能量
沿着箭头方向(X轴负方向)加不同外电场(-0.02—0.07 a.u.),采用DFT/B3LYP方法6-31G(d)基组进行结构优化,得到分子稳定构型. 不同外电场下的氟氯碳酰分子键长的变化如图2所示,分子C-O和C-Cl键长随外电场增强而增大,C-F键长随外电场增强而减小;分子键长随外电场的变化规律可以用分子内部电场的变化来解释,当有外电场存在时,分子内应力和外电场力的合力决定着外电场下分子的稳定结构[33];总体来看,C-Cl键能量最高,且外加电场越大,C-Cl键能量越高,即C、Cl核间距增大,C-Cl键越容易断裂[34]. C和Cl原子间净电荷量如表2所示,随外电场强度净电荷量均逐渐增强,说明C、CL原子间库仑引力增大,C-Cl键长增大;而C、F原子间反之.
不同外电场下分子总能量和电偶极矩的变化如图3所示,总能量随外电场增强先增大后减小,而电偶极矩随外电场增强先减小后增大;分子体系和外电场的相互作用能为Hint=-μF,而电偶极矩的增大,使分子体系与外电场的相互作用能减小,这也可以解释分子体系的总能量随外电场的增强而减小的现象[4].
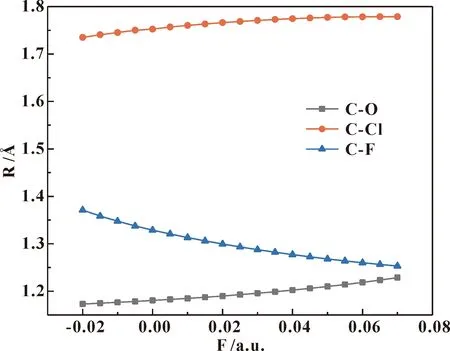
图2 不同外电场下的氟氯碳酰分子键长变化图Fig.2 The changes of carbonyl chloride fluoride molecular bond length under different external electric fields
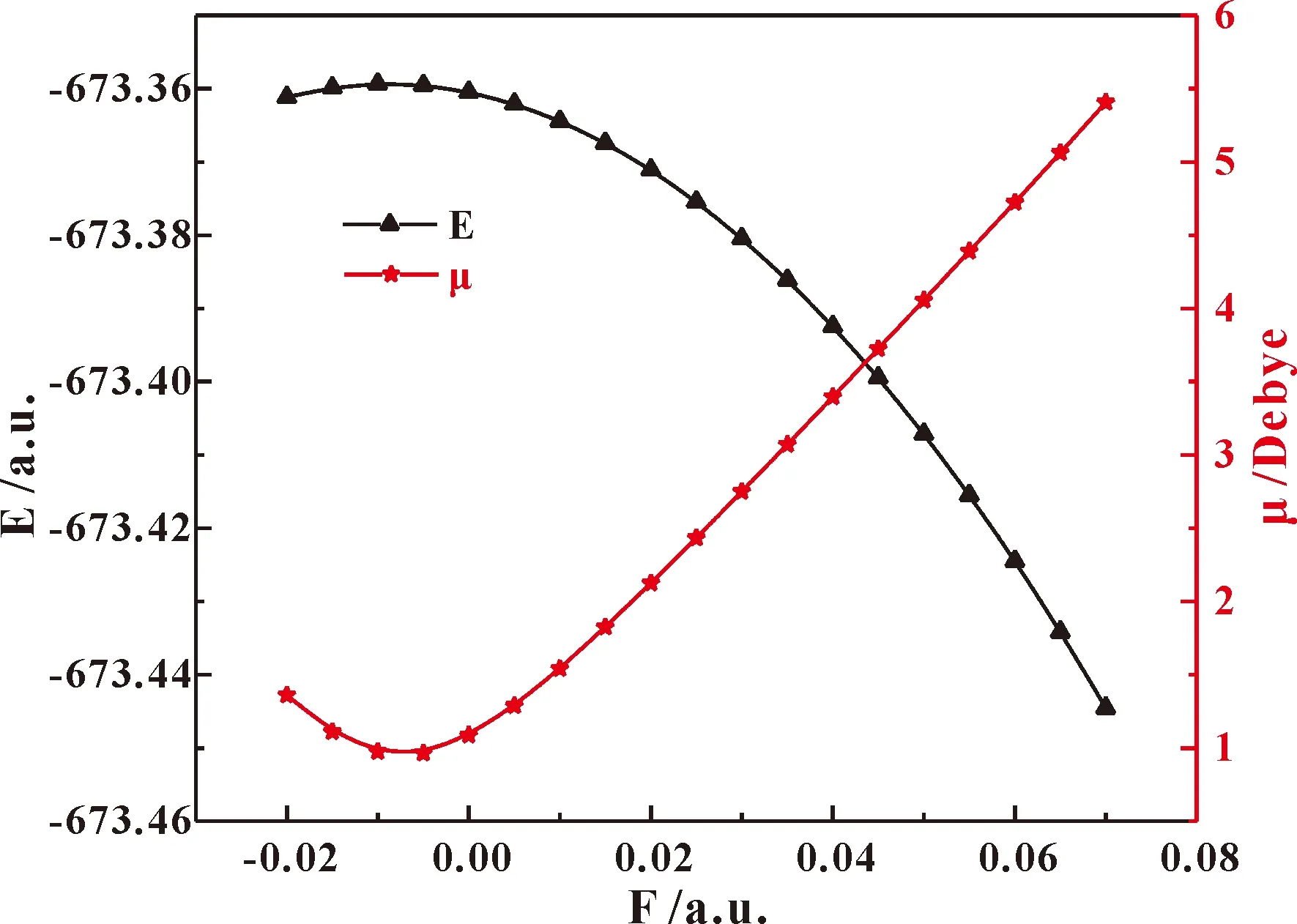
图3 不同外电场下的氟氯碳酰分子总能量、偶极矩变化图Fig.3 The changes of carbonyl chloride fluoride molecular total energy and dipole moment under different external electric fields
3.3 外加电场下氟氯碳酰的分子轨道能级和能隙
不同外电场(-0.02—0.07a.u.)作用下,分子的最低空轨道(LUMO)能量EL和最高占据轨道(HOMO)能量EH以及能隙EG的影响如表2所示,其中能隙EG计算公式为:
EG=(EL-EH)×27.2eV
(3)
EL表示分子得到电子能力的强弱,EL越小越容易得到电子;EH表示分子失去电子能力的强弱,EH越大越容易失去电子;EG表示分子参加化学反应的能力,EG越小分子越容易被激发到激发态而发生化学反应[34]. 从图4可以看出,随着外电场的增强EL逐渐减小,EH也显示减小趋势,但EL减小趋势比EH大,因此,能隙的变化如图4所示,随着外电场增强EG呈减小趋势,说明电子越容易被激发到空轨道而形成空穴子,从而发生化学反应[5],与上文结论一致.
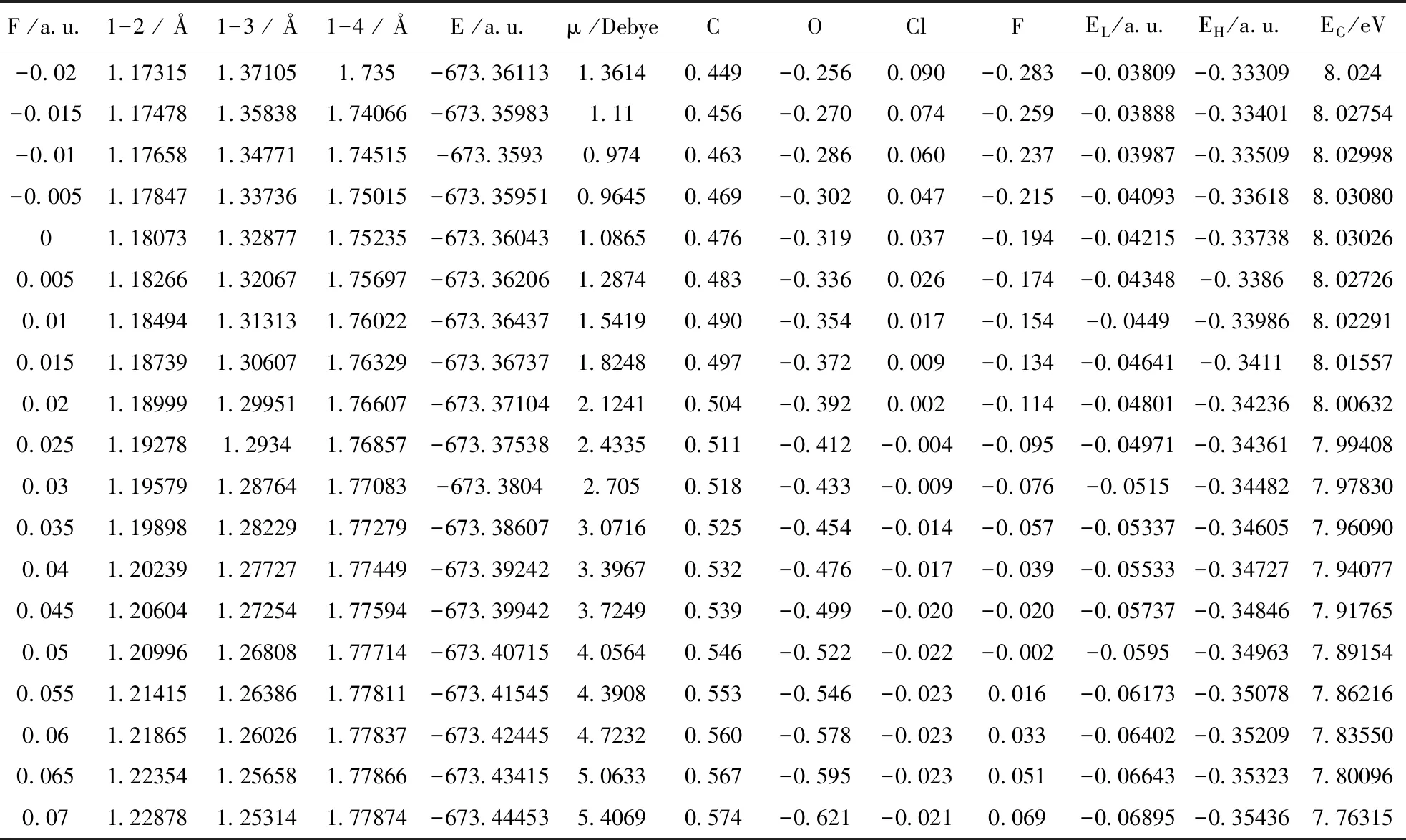
表2 不同外电场下氟氯碳酰分子几何结构

图4 不同外电场作用下氟氯碳酰分子的最低空轨道能、最高占据轨道能和能隙变化图Fig.4 The changes of the lowest empty orbital energy, the highest occupied orbital energy and energy gap for carbonyl chloride fluoride molecule under different external electric fields
3.4 外加电场下氟氯碳酰分子的振动频率、红外光谱及拉曼光谱
采用DFT/B3LYP方法6-31G(d)基组对氟氯碳酰分子结构进行优化,研究了不同外电场(-0.02—0.07 a.u.)作用下,对分子振动频率、红外光谱和拉曼光谱的影响. 先计算零电场下振动模型数据与实验值进行对比,计算值与实验值符合较好,如表3所示,进一步说明了计算的正确性.
如图5是不同外电场下分子红外光谱图,从图中可以看出该分子有6种振动模型,与实验得到的相匹配. 图5a主要展示了分子CF stretch(str)、CO str两种最强振动,随外电场增强CF str振动发生蓝移,而CO str振动发生了红移;为更清楚观察其规律,将图5a 675—875 cm-1范围放大如图5b所示,从图中可以看出CCl str、O p-deform两种振动,随外电场增强CCl str、O p-deform振动均发生蓝移,且振动强度都逐渐增大,但CCl str振动比O p-deform振动受影响更大;图c是400—530 cm-1范围内的放大图,图中主要展现了CClF deform、CO deform两种振动,随外电场增强CClF deform振动并没有发生移动,只是强度增强,而CO deform振动先红后蓝,强度先增强后减小.
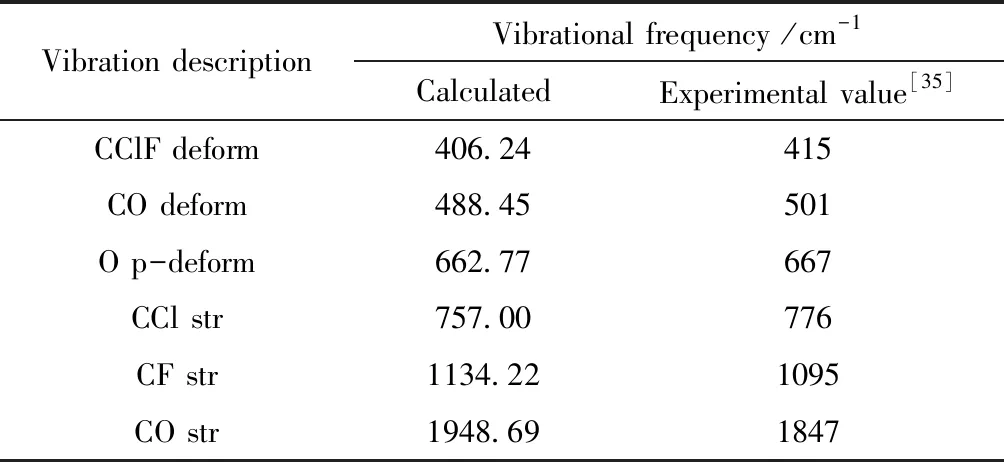
表3 红外光谱的计算值与实验值
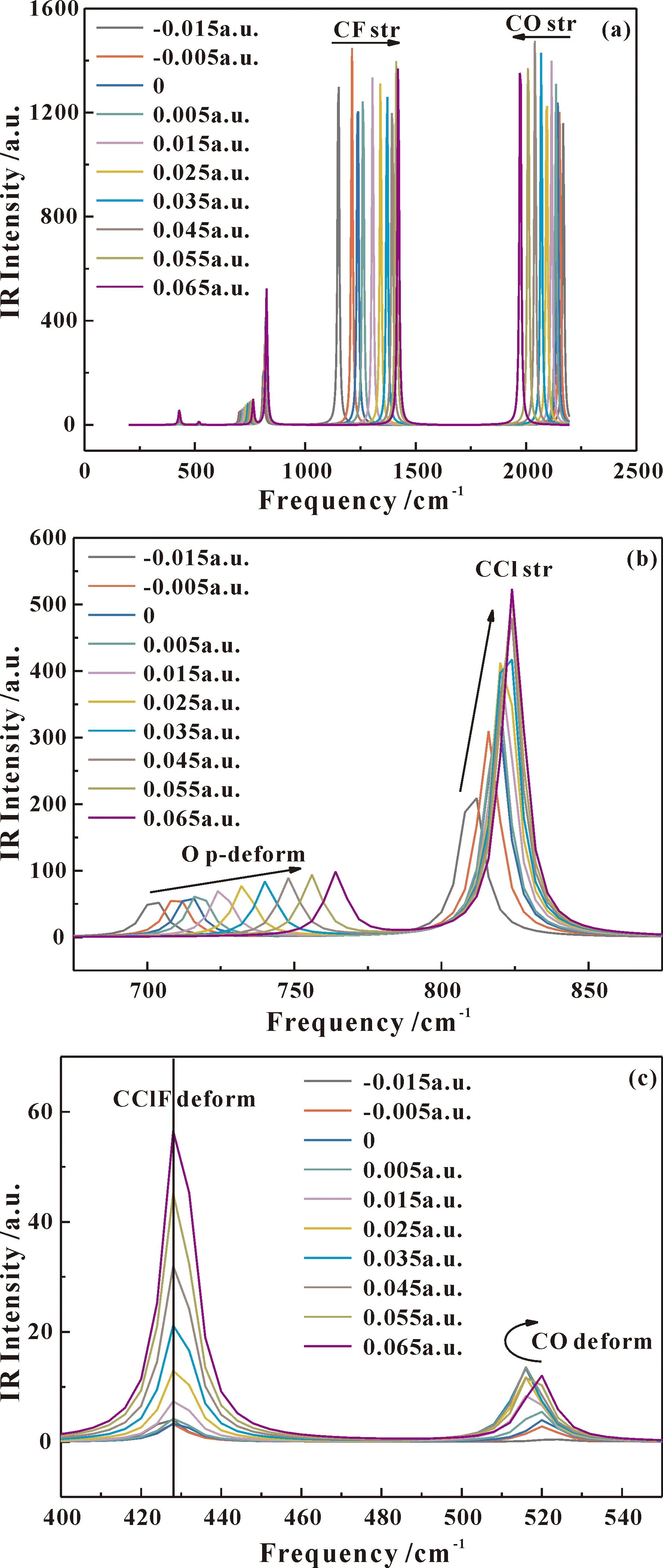
图5 不同外电场作用下氟氯碳酰分子红外光谱变化图Fig.5 The changes of carbonyl chloride fluoride molecular infrared spectrum under different external electric fields
如图6是不同外电场下分子拉曼光谱图,从图6a中可以看出该分子有CClF deform、CO deform、O p-deform、CCl str、CF str、CO str 6种振动模型,与红外光谱相一致. 从图6b可以看出,外加电场作用下,CClF deform和CO deform振动的影响较小,没有发生移动;O p-deform、CCl str、CF str均发生了红移,但是CCl str的红移幅度很小;CO str发生了蓝移现象,且振动幅度逐渐减小.
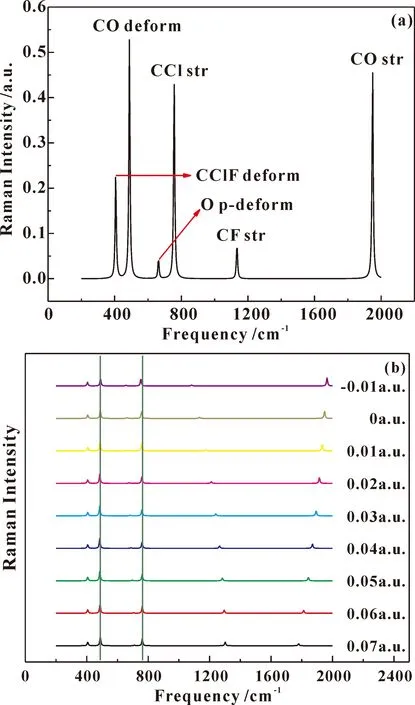
图6 不同外电场作用下氟氯碳酰分子拉曼光谱变化图Fig.6 The changes of carbonyl chloride fluoride molecular Raman spectrum under different external electric fields
3.5 外加电场下氟氯碳酰分子的解离能
3.5.1外加电场下氟氯碳酰分子的C-Cl键键长
在无外电场和不同外电场(-0.015—0.03 a.u.)作用下,能量扫描计算氟氯碳酰基态分子,得到C-Cl键束缚势能曲线,其变化如图7所示. 沿着图1所示方向加外电场时,随着外电场的增强,氟氯碳酰分子的势能面逐渐降低,但降低幅度较小;即电场越强氟氯碳酰分子的C-Cl键越容易断裂,分子越容易解离;当外电场强度为0.03 a.u.时,C-Cl键势垒消失,分子应该发生了解离.
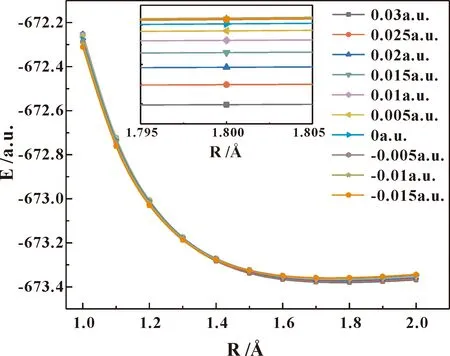
图7 在不同外电场作用下氟氯碳酰分子C-Cl键解离势能曲线Fig.7 The changes of the potential energy surface of the C-Cl bond dissociation of carbonyl chloride fluoride molecules under different external electric fields
3.5.1外加电场下氟氯碳酰分子的逐步解离
在无外电场和施加不同外电场(-0.015—0.03 a.u.)作用下,采用上述方法和基组对氟氯碳酰基态分子的C-F和C-Cl键长进行了扫描,C-F和C-Cl键均为1 Å-2 Å,得到如图8所示的势能曲面,其中红色为0.03 a.u.电场下的势能面,黑色为零电场下的势能面,绿色为-0.05 a.u.电场下的势能面. 在零点场下,分子处于束缚态. 加外电场时,随着外电场的降低,C-Cl键发生断裂,且势能面逐渐降低,C-F键的势垒也逐渐降低,即C-Cl键发生断裂, C-F键也逐渐断裂,很有可能发生了逐步解离.
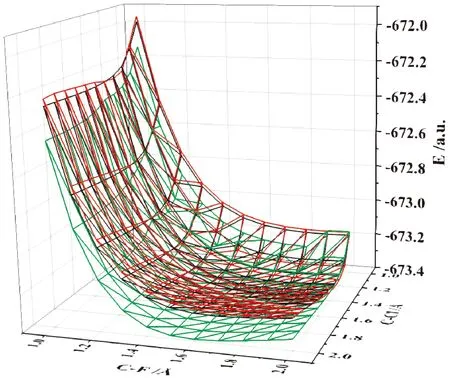
图8 在不同外电场作用下氟氯碳酰分子解离势能面Fig. 8 Dissociation potential energy surfaces of carbonyl chloride fluoride molecules under different external electric fields
4 结 论
采用密度泛函理论在B3LYP/6-31G(d)水平上加不同外电场,研究氟氯碳酰分子物理性质的变化规律. 研究表明,在外电场作用下分子结构变化比较明显,沿X轴负方向加外电场(-0.02—0.07 a.u.)时,分子C-O键长、C-Cl键长随外电场的增强而逐渐增大,C-F键长随外电场的增强而减小;分子体系的总能量随外电场的增强而减小,偶极矩随外电场的增强而增大;能隙EG随外电场的增强先增大后减小,同时分子6种振动形式中,CClF deform振动的红外光谱和拉曼光谱均没有发生移动,CO deform振动的拉曼光谱没有发生移动,但是红外光谱先红移后蓝移,O p-deform、CCl str、CF str振动的红外光谱发生了蓝移,拉曼光谱红移了,但是CCl str的拉曼光谱蓝移非常微弱,CO str振动的红外光谱发生了红移,而拉曼光谱发生蓝移现象. 随着外电场增强,分子的势能曲线逐渐降低,即分子越容易发生解离,当外电场强度为0.03 a.u.时,分子的势垒消失,分子发生解离;外电场为-0.005 a.u.时,分子两个键断裂,发生逐步解离. 本研究可对氟氯碳酰分子进行外电场降解、收集、并计算得到基态几何数据,并尝试观察分子的协同解离现象,这将为进一步研究氟氯碳酰分子提供理论依据.
