大豆球蛋白A1a亚基同源建模及抗原表位的预测
2022-05-09付杨席俊陈慧彬陈阳
付杨,席俊,陈慧彬,陈阳
(河南工业大学 粮油食品学院,河南 郑州,450001)
大豆起源于亚洲,是一种被大面积种植的油料作物,蛋白质含量约占大豆总质量的40%[1]。大豆富含异黄酮和叶酸,能对人体健康产生有益的影响[2],而被加工成各类食品,在全球范围内被广泛食用。但大豆也是最容易导致人体过敏的八大食品之一[3],过敏患者会出现荨麻疹、哮喘、肠道综合征等反应,严重时还会造成休克威胁生命[4]。HELM等[5]发现大豆中独特的蛋白质可能是大豆过敏反应的原因,尤其是大豆球蛋白和β-伴大豆球蛋白。大豆球蛋白致敏性主要取决于其单体组成亚基的过敏性,大豆球蛋白亚基在还原条件下可以解离为一条酸性多肽链A(30~45 kDa)和一条碱性多肽链B(18~20 kDa)[6],大豆球蛋白A1a(Glycinin A1a)是大豆球蛋白G1亚基中的酸性多肽,DJURTOFT等[7]也证明免疫球蛋白E可以和所有的酸性亚基结合,现有报道表明大豆蛋白的致敏性源自于其酸性亚基,但Glycinin A1a亚基的具体线性B细胞表位未见报道。本实验是对Glycinin A1a亚基上最有可能的B细胞表位进行预测。
过敏反应的产生要依靠免疫细胞对抗原分子的特定部位即抗原决定簇完成识别[8],作为抗原中的线性片段或构象结构,B细胞表位能被抗体特异性识别,因此成为表位预测的重要部分。对于抗原表位的鉴定、预测,李俊慧等[9]总结了一些现有常用生物信息学方法和软件。利用生物信息学软件对蛋白组学和基因组学的原件进行模型建立,同时以数据库中已收录的各种抗原信息为依据,对待研究的过敏原线性表位进行筛选和预测。最后使用生物学知识和免疫学实验来确定预测的表位是否具有功能性,这种多学科交叉的方法,不仅节约大量的人力物力,加快实验进程,而且拓展了表位预测的技术平台,为预测更多未知的抗原表位提供了可能。刘阳星月等[10]通过蛋白质数据库(protein data bank,PDB)和系列生物预测软件、在线网站成功预测出大豆过敏原11S球蛋白G2中A2链的结合表位。闫慧丽等[8]利用SWISS-MODEL和Deep View 4.1完成了大豆主要过敏原Gly m Bd 28K的同源建模和评估,预测出6个构象表位相关区段。皮江一等[11]也凭借该方法预测出了大豆主要过敏原β-伴大豆球蛋白β亚基的抗原表位,并以此为基础对预测表位区域进行了分段克隆,抗原表位的预测成为后续抗原位置鉴定的重要前提。本研究以美国国家生物信息技术中心(National Center of Biotechnology Information,NCBI)数据库中得到的Glycinin A1a亚基氨基酸序列为原材料,以Glycinin A1a的高同源性蛋白质结构为模板,生成Glycinin A1a三维模型。用软件DNAStar、SOPMA、ABCpred和BepiPred 1.0 server、DiscoTope 2.0 server分别进行线性表位和构象表位的预测。为后期大豆球蛋白免疫检测提供理论参考;准确定位大豆球蛋白亚基的过敏位点,就可以有针对性的对过敏位点进行加工处理,为开发降低、消除大豆过敏性的食品加工技术提供帮助。
1 材料与方法
1.1 材料
使用NCBI GenBank数据库查找出Glycinin A1a亚基所对应的氨基酸序列。该序列共有287个氨基酸排列如下:
FSSREQPQQNECQIQKLNALKPDNRIESEGGLIE-TWNPNNKPFQCAGVALSRCTLNRNALRRPSYTNGPQIYIQQGKGIFGMIYPGCPSTFEEPQQPQQRGQSS-RPQDRHQKIYNFREGDLIAVPTGVAWWMYNNEDT PVVAVSIIDTNSLENQLDQMPRRFYLAGNQEQE-FLKYQQEQGGHQSQKGKHQQEEENEGGSILSGFTL-EFLEHAFSVDKQIAKNLQGENEGEDKGAIVTVKGGL-SVIKPPTDEQQQRPQEEEEEEEDEKPQCKGKDKHCQ-RPRGSQSK
查找同源蛋白的PDB在线网站;用于三级结构模型建立和模型稳定性分析的软件SWISS-MODEL和Deep View 4.1;DNAStar软件包中的子程序Protean;其他预测软件:SOPMA、ABCpred、BePiPred 1.0 server和DiscoTope 2.0 server。
1.2 实验方法
1.2.1 Glycinin A1a亚基线性表位预测
DNAStar中的Protean程序可以对氨基酸序列进行分析,包括疏水性、柔韧性、抗原指数、表面可及性[12]。因为所测指标与抗原表位密切相关,准确性也更强,具有较高的参考价值。
SOPMA是在线网站预测工具,打开网站页面直接将Glycinin A1a亚基氨基酸序列以FASTA格式粘贴在序列框内,选用默认的预测参数,提交序列可得到结果。
BepiPred 1.0预测服务器基于隐马尔可夫模型的组合和倾向量表方法对B细胞表位预测[13]。操作方法与SOPMA相类似。
ABCpred所预测的表位氨基酸数量是相等的,排列的顺序是根据表位的可能性大小,预测准确性相对其他软件较低,作为参考。使用参数参照刘阳星月等[10]的方法。
1.2.2 Glycinin A1a亚基同源建模及三维模型稳定性评估
根据Glycinin A1a亚基的氨基酸序列在PDB网站上查找出相应的同源蛋白,以同源性最高的蛋白作为模板[14],将两者的氨基酸序列都输入SWISS-MODEL的序列框中,采用Target-Template Alignment模式建立模型,建立的三维模型保存为PDB文件。打开Deep View4.1软件,在“File”选项下打开模型PDB文件;在工具栏“wind”选项中点击“Ramachandran Plot”即可得到建立模型的拉氏图。
1.2.3 GlycininA1a亚基构象表位预测
使用DiscoTope 2.0 Server在线软件,选择导入的文件为同源建模保存的模型PDB文件,参数使用默认值-3.7,提交文件即可预测。
2 结果与分析
2.1 Glycinin A1a亚基的线性表位预测结果
2.1.1 DNAStar
使用DNAStar中的子程序Protean预测Glycinin A1a亚基的亲水性、柔韧性、抗原指数、表面可及性如图1所示。由于抗原位点是那些被抗体识别的位点,这些位点很可能是容易接触或在蛋白质表面的,这些区域要比内部区域更具流动性,由此推测它们是亲水的,亲水性越强,越容易和抗原表位结合。蛋白氨基酸残基多数处于亲水区,且分布均匀。柔韧性体现在图1中蓝色矩形覆盖区,柔韧性和表面可及性较强时易于折叠、弯曲,能够促进二级结构的形成可作为预测的重点区段[15]。抗原指数是直接就其抗原可能性预测,选择抗原指数>0的区域作为预测的表位。预测时将这4种指标中可能性较高区段进行综合比对,同时符合4种指标要求的序列,更有机会被视为表位。以上预测结果如表1所示。

图1 DNAStar分析结果Fig.1 DNAStar analysis results
2.1.2 SOPMA
SOPMA可以计算出蛋白质二级结构中的α-螺旋、β-折叠、β-转角和无规则卷曲所占的百分比,如图2所示。Glycinin A1a亚基中无规则卷曲氨基酸残基含量最高占50.52%,α-螺旋是由24.74%氨基酸残基构成,β-转角和β-折叠所含的氨基酸残基分别占16.72%和8.01%。在蛋白质的二级结构中α螺旋和β折叠一般不作为抗原表位[16-19],因为氢键的存在结构稳定不易变形,而且α螺旋和β折叠通常位于蛋白内部,不易与受体接触。而β-转角和无规则卷曲一般位于蛋白质表面,为了满足蛋白质的功能需要,表面结构必须进行适当的改变也更容易被识别与抗原表位结合,是致敏表位的几率更大。由此结果预测的表位如表2所示。

表1 DNAStar 预测的抗原表位Table 1 Predicted antigen epitope by DNAStar
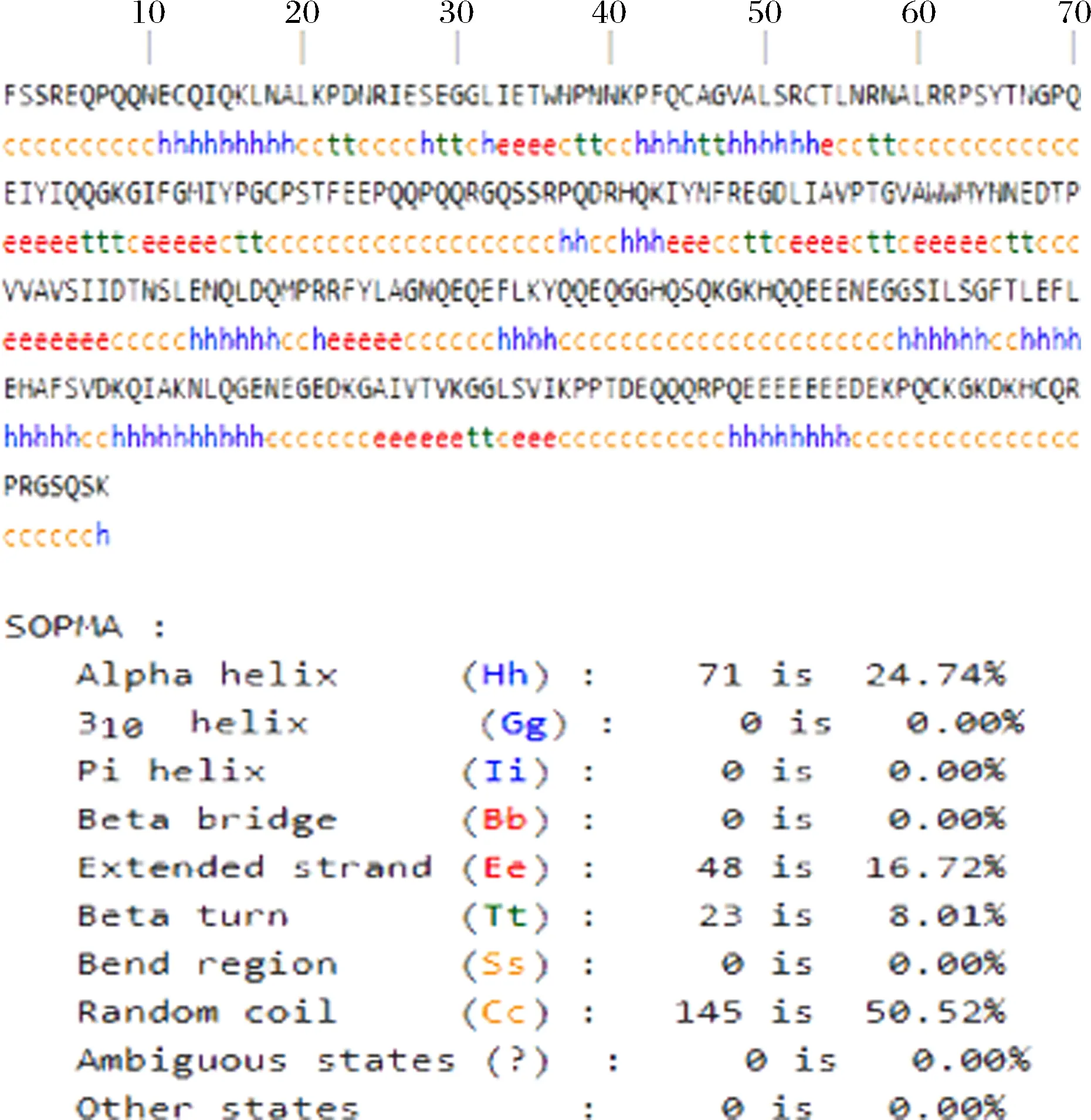
图2 SOPMA分析的大豆球蛋白A1a二级结构结果Fig.2 Secondary structure of the Glycinin A1a analyzed by SOPMA
2.1.3 BepiPred
BepiPred 1.0在线网站预测表位得到的结果如表3所示。序列段长度差别较大,但得到的表位分布较为均匀,而且序列段与DNAStar和SOPMA预测的表位位置区域高度重合,表明预测结果具有较高可信度。

表2 SOPMA 预测的Glycinin A1a抗原表位Table 2 Predicted antigen epitope by SOPMA

表3 BepriPred预测的Glycinin A1a抗原表位Table 3 Predicted antigen epitope by BepriPred
2.1.4 ABC pripred
ABC pripred对Glycinin A1a亚基预测了18个线性表位,按照可能性大小如表4排列,最有可能的线性表位处于氨基酸序列的24~33位,在综合评价时更侧重于排名前4的预测表位。通过分析发现排名靠前的表位大部分靠近蛋白的N端。

表4 ABC pripred预测的Glycinin A1a抗原表位Table 4 Predicted antigen epitope by ABC pripred
线性构象表位预测结果,按照刘阳星月等[10]提出的方法对4种软件预测结果进行整合,结果如图3所示,预测出10个可能的Glycinin A1a抗原线性表位EQPQQN、KPDNRI、NPNNK、RRPSYTNG、PQQPQQRGQS、QEQGGHQSQKGKHQQEEENE、NEGED、PPTDEQQQRP、DEKPQCKGK、RPRGS。
2.2 Glycinin A1a亚基模型的建立和稳定性评估
同源建模是按未知结构蛋白氨基酸序列在蛋白质结构数据库中查找出已有的相似的蛋白质的结构,然后将此蛋白结构进行优化,进而建立出未知结构蛋白的模型[14]。SWISS-Model对大豆球蛋白A1a亚基建立模型找出大豆球蛋白A1a亚基的同源蛋白,结果显示1FXZ(大豆球蛋白A1aB1b同源三聚体)与Glycinin A1a亚基同源性高达71%。以1FXZ为模板对大豆球蛋白A1a亚基完成同源建模,模型显示如图4所示。此模型符合大豆蛋白六聚体复合物结构。利用Deep view 4.1对建立出来的Glycinin A1a亚基模型进行稳定性分析,得到的拉氏图如图5所示。

图4 Glycinin A1a亚基预测模型Fig.4 Prediction model of the Glycinin A1a subunits

图5 Glycinin A1a亚基Ramachandran plotFig.5 Ramachandran plot of the Glycinin A1a subunits
以Φ和Ψ为坐标轴所组成的二维图,来表示α碳原子和肽平面间单键的旋转形成的两面角[20],并不是所有Φ和Ψ形成的角都适合多肽骨架,不同的氨基酸残基能够稳定存在时对Φ和Ψ值是有范围的,根据这个范围可将二维图分为允许区(黄色线内区域)、最大允许区(蓝色色线内区域)和不允许区[21]。由图5计算得到Glycinin A1a亚基的氨基酸残基超过90%落在允许区内,即建立的A1a亚基模型是可以稳定存在的。
2.3 Glycinin A1a亚基构象表位分布
使用PyMOL软件将DiscoTope 2.0 Server预测的Glycinin A1a亚基构象表位在模型中表示出来如图6所示。发现预测的构象表位均处于模型的表面,而且较多表位处在无规则卷曲处,符合李雪娇等[15]的实验结果,也正是无规则卷曲部位的独特结构使得预测的线性表位和构象表位有部分重合。经过对线性表位和构象表位的对比发现:两者虽然不是完全相同的,但是在线性表位当中包含了部分构象表位。表明预测结果符合已有研究:线性表位和构象表位之间是密切相关的[22]。

a-正面;b-反面图6 Glycinin A1a亚基构象表位Fig.6 Glycinin A1a sub-base conformation epitope 注:红色部分表示氨基酸数量>5个的表位,黄色部分表示 氨基酸数量≤5个的表位
3 结论
抗原表位的预测方法多种多样,但都是基于蛋白质的结构组成和物理化学性质,在本研究中DNAStar分析了Glycinin A1a亚基的亲水性、柔韧性、表面可及性、抗原指数,因为这些性质的不同是抗原表位区别于普通位点的关键。由于α螺旋和β折叠结构不易改变且大多位于蛋白内部难以与抗体接触结合,不作为表位处理,相反,β转角和无规则卷曲凭借良好的柔韧性、处于表面的优势可以较好的被识别,也成为表位预测的重要指标。最终得到了10个可能性较高的线性表位:24EQPQQN29、40KPDNRI45、56NPNNK60、80RRPSYTNG87、114PQQPQQRGQS123、197QEQGGHQSQKGKHQQEEENE216、247NEGED251、267PPTDEQQQRP276、285DEKPQCKGK293、299RPRGS303。本研究还对Glycinin A1a进行了构象表位的预测。预测结果与SAEED等[23]的实验结果相符合,即抗原构象表位位于3D模型的表面。符合一般表位预测原则,处于表面的位点可以更好的与抗体嵌合。
此实验为接下来的Glycinin A1a过敏性表位的筛选和鉴定提供了数据支持,能更有针对性的去验证各个表位的免疫原性。也为改善加工条件,降低大豆致敏性提供理论支持。此研究借助机器预测抗原表位,避免了大量时间,人力和湿实验的耗费[24]。更重要的是并在没有人为干预的情况下对未知数据进行预测。大大降低了实验的误差。
