高效液相色谱法测定腰痛丸中蛇床子素、升麻素苷和橙皮苷的含量
2022-05-07仇其原朱干红
仇其原,张 卓,朱干红
1 盐城市食品药品监督检验中心,盐城 224000;2 盐城市中医院,盐城 224000
腰痛丸(Ⅰ)为棕褐色至棕黑色的水蜜丸;气微香,味稍微苦,甘,辛。其由15 味中药配伍组成,方中独活、防风、防己、威灵仙、续断、制川乌、制草乌、苍术为君药,主祛风散寒除湿;干姜、肉桂以温中散寒、通络止痛,乌梢蛇、蜈蚣以祛风通络、解痉镇痛共为臣药;茯苓、陈皮以利水渗湿、健脾益胃为佐药;甘草为使药,调和诸药,并降低制川乌、制草乌的毒性。该药主治祛风散寒、通络止痛,风寒腰痛等症。
腰痛丸(Ⅰ)中,独活、防风、陈皮的有效成分分别为蛇床子素[1]、升麻素苷[2]、橙皮苷[3]。相关药理学试验表明,蛇床子素、升麻素苷和橙皮苷是腰痛丸(Ⅰ)发挥药效作用的主要的活性成分,建立这3 种有效成分(结构式见图1)的含量测定方法,对于增加腰痛丸(Ⅰ)的药效物质基础的质量研究具有积极的作用[4]。
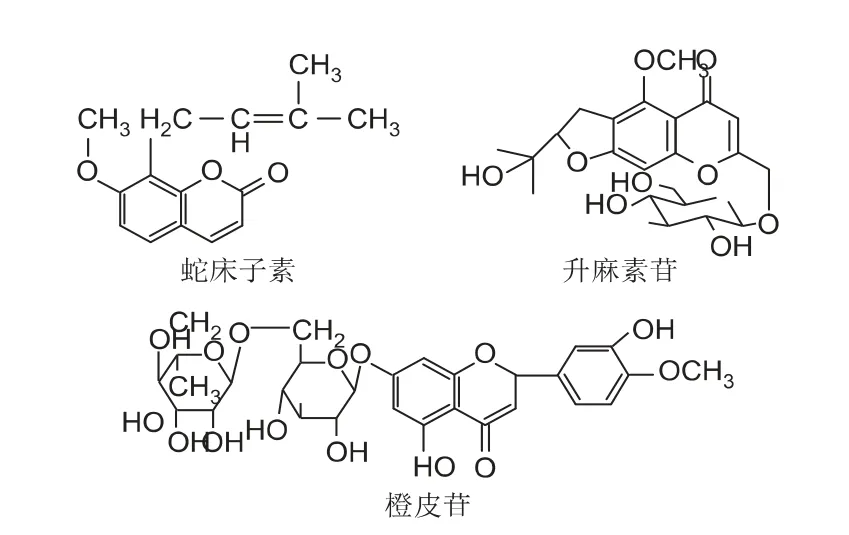
图1 腰痛丸(Ⅰ)中独活、防风、陈皮的有效成分及结构式
中药复方制剂的成分复杂,仅凭单味药的定性方法并不能全面有效控制腰痛丸(Ⅰ)质量与疗效的一致性。本实验采用HPLC 法,以腰痛丸(Ⅰ)中独活、防风、陈皮的各有效成分蛇床子素、升麻素苷、橙皮苷作为指标性成分,建立同时测定其含量的方法,并完成方法学研究,以可靠的方法及数据依据[3],为建立腰痛丸(Ⅰ)制剂质量的标准提供可行性方案。
1 仪器与药品、试剂
仪器:Waters2695 高效液相色谱仪、Waters2996二极管阵列紫外检测器(PAD),Empower 色谱工作站(美国Waters 公司);XS205 DU 电子天平(瑞士梅特勒-托利多公司)。
对照品:蛇床子素(批号110822-200407)、升麻素苷(批号111522-201209)、橙皮苷(批号110721-201316,110721-201115)均购自中国食品药品检定研究院。供试品:腰痛丸(Ⅰ)(盐城市中医院制剂室,批号190115、190601、190907)。试剂:甲醇、醋酸铵溶液为化学纯;水为纯化水。
2 方法与结果
2.1 溶液的制备
2.1.1 混合对照品溶液精密称取蛇床子素对照品15 mg,置于100 mL 量瓶中,加入甲醇溶解并稀释至刻度,混匀,即得对照品溶液(1);精密称取升麻素苷对照品15 mg,置于10 mL 量瓶中,加甲醇溶解并稀释至刻度,混匀,即得对照品溶液(2);精密称取橙皮苷对照品15 mg,置于20 mL 量瓶中,加甲醇溶解并稀释至刻度,混匀,即得对照品溶液(3)。依次从对照品溶液(1)、(2)、(3)中精密吸取1、1、2 mL,置于同一20 mL 量瓶中,加甲醇稀释至刻度,混匀,即得混合对照品溶液①。同法依次从对照品溶液(1)、(2)、(3)中精密吸取2、2、4 mL,置于同一10 mL量瓶中,加甲醇稀释至刻度,混匀,即得混合对照品溶液②。
2.1.2 供试品溶液从3 个批次中随机称取腰痛丸(Ⅰ)适量,研细,取约5 g,精密称定,置于具塞锥形瓶中,加甲醇100 mL,称重,水浴回流2 h,放冷,称定重量,加甲醇补到原先的重量,摇匀,滤过,即得。
2.2 色谱条件
色谱柱:Waters CAPCELL PAK C18(4.6 mm×250 mm,5 μm);流动相:0.2 mol·L-1醋酸铵溶液(A)-甲醇(B),梯度洗脱程序为0 min 80%A,20B;30 min 35A,65B;50 min 35A,65B;检测波长:284 nm(升麻素苷、橙皮苷),324 nm(蛇床子素);柱温:30 ℃;流速:1.0 mL·min-1;进样量:20 μL。系统适用性要求:供试品色谱图中,升麻素苷、橙皮苷、蛇床子素与其他峰的分离度大于1.5,理论塔板数不低于8000。
2.3 方法学考察
2.3.1 专属性试验以“2.1”项下方法所得供试品溶液及混合对照品溶液,按“2.2”项下色谱条件进行专属性试验。见图2。经全波长扫描图谱的对比,供试品中蛇床子素、升麻素苷和橙皮苷峰与各对照品峰均得到指认,并达到基线分离,分离度良好,符合专属性及系统适用性要求。
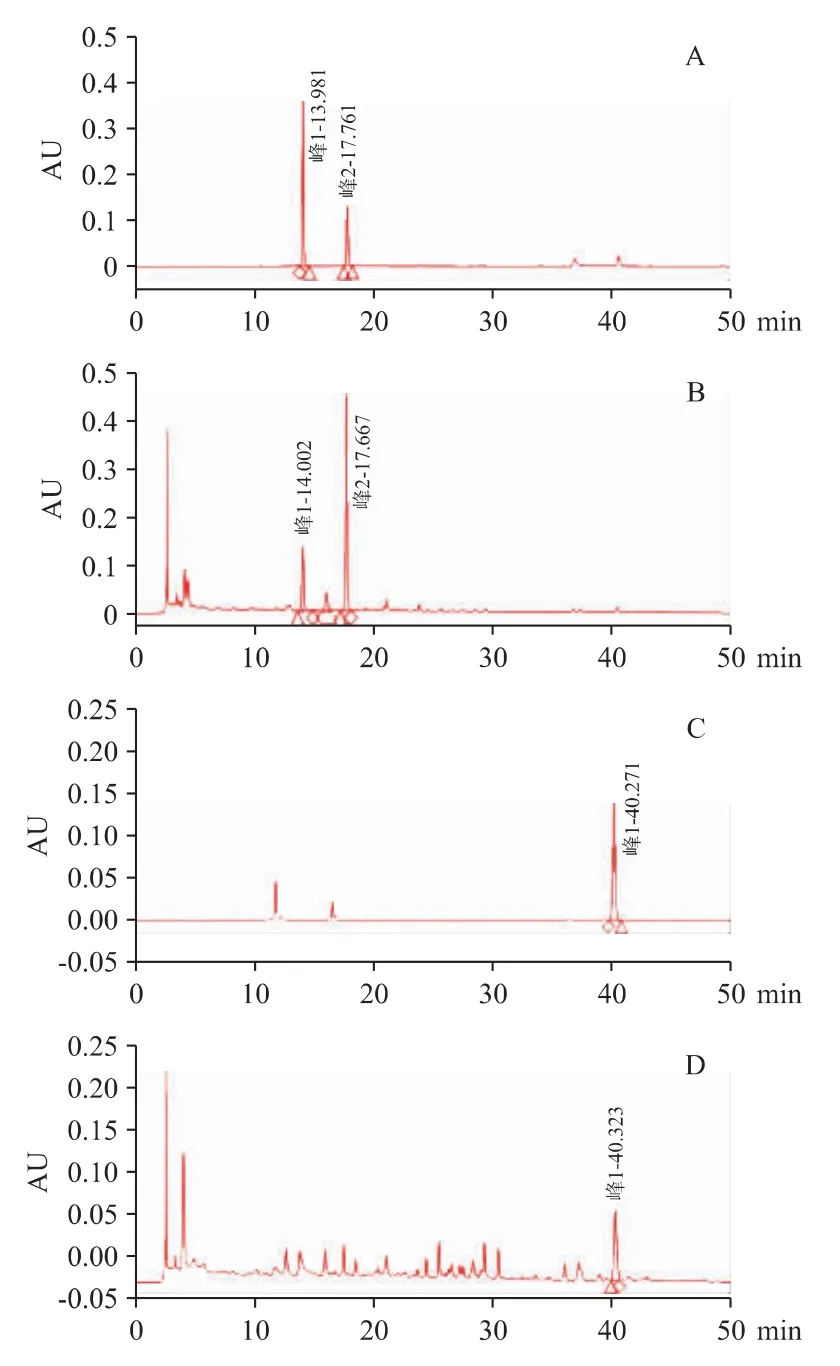
图2 腰痛丸(Ⅰ)专属性试验色谱图
2.3.2 线性与范围依次从对照品溶液(1)、(2)、(3)中精密移取适量制成线性溶液,其中蛇床子素的梯度浓度为3.8、7.5、15.0、22.5、30.0 μg·mL-1;升麻素苷及橙皮苷的梯度浓度均为38、75、150、225、300 μg·mL-1;通过“2.2”项下色谱条件进样,以线性溶液中各成分相应的浓度C(μg·mL-1)为横坐标,峰面积Y(mAu*s)为纵坐标,经线性回归分别得回归方程:蛇床子素Y=5.67×105C+2.22×104,r2=0.996 0;升麻素苷Y=2.00×106C+2.30×104,r2=0.998 0;橙皮苷Y=1.00×106C+1.05×105,r2=0.998 0。在相应的线性范畴内,3 成分均呈良好的线性关系。
2.3.3 进样精密度试验取“2.1”项下所配供试品溶液及混合对照品溶液适量,按“2.2”项下色谱条件连续进样6 次,分别计算各指标性成分峰面积的相对标准偏差(RSD),蛇床子素、升麻素苷、橙皮苷峰面积RSD分别为0.44%、0.94%、0.32%。表明仪器精密度良好。
2.3.4 稳定性试验取同一批号的腰痛丸(Ⅰ)制备供试品溶液,于0、2、4、8、16、24 h 按“2.2”项下色谱条件进样测定,得供试品溶液中蛇床子素、升麻素苷、橙皮苷的峰面积的RSD 分别为:1.06%、0.93%、0.46%,表明供试品溶液24 h 内有良好的稳定性。
2.3.5 重复性考察取同一批号腰痛丸(Ⅰ)平行制备6 份供试品溶液,按“2.2”项下色谱条件进样测定,按标准曲线法分别计算各指标性成分含量的RSD 值。结果蛇床子素、升麻素苷、橙皮苷的含量RSD 分别为0.91%、0.74%、0.13%,表明本方法重复性良好。
2.3.6 加样回收率试验精密称取已知含量的腰痛丸(Ⅰ)5g,共5 份,置于具塞瓶中,精密加入蛇床子素对照品溶液(0.5 mg·mL-1)1.0 mL,升麻素苷对照品溶液(3.0 mg·mL-1)1.0 mL,橙皮苷对照品溶液(10 mg·mL-1)1.0 mL,加甲醇100 mL,称重,水浴回流2 h,放冷,称重,加甲醇补到原先的重量,摇匀,滤过,即得加样回收率试验的供试品溶液。按“2.2”项下色谱条件进样测定,并根据公式:回收率=(实测量-供试品所含被测成分量)/加入对照品量×100%,计算回收率,蛇床子素、升麻素苷、橙皮苷的加样回收率分别为98.43%、98.20%、98.50%,可以满足实验要求。
2.4 样品含量测定
取3 个批号腰痛丸(Ⅰ)样品适量,共2 份,依照以上方法配制供试品溶液,按“2.2”项下色谱条件进行含量测定,平均含量结果见表1。表明所建立的高效液相色谱方法检测腰痛丸(Ⅰ)中蛇床子素、升麻素苷以及橙皮苷的含量准确可行。
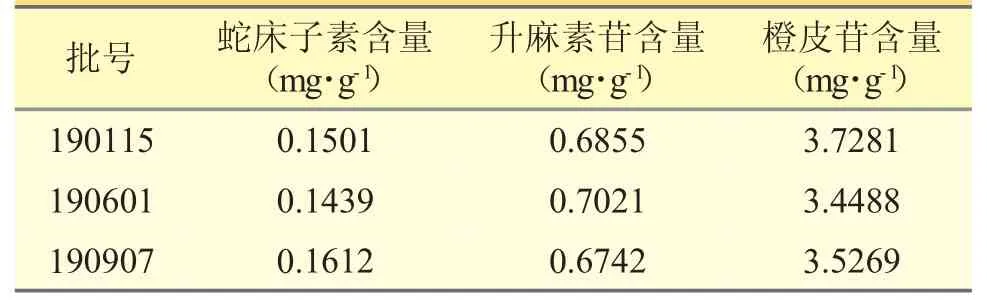
表1 样品含量测定(n=2)
3 讨论
3.1 流动相选择
在流动相的选择中,曾考察不同比例的甲醇-0.2 mol·L-1醋酸铵溶液系统和甲醇-水系统作为流动相进行梯度洗脱,结果发现,以前者作为流动相进行梯度洗脱时,所得蛇床子素、升麻素苷和橙皮苷各组分的峰形良好,且3 种组分分离度都符合要求。
3.2 检测波长的选择
本实验参考2020 版《中国药典》[5]中蛇床子素、升麻素苷和橙皮苷的检测波长,分别进行全波长扫描,结果升麻素苷和橙皮苷在波长284 nm 处均有较大吸收;另参考相关文献,蛇床子素在波长324 nm处有最大吸收[4]。故分别确定之。
4 小 结
建立了HPLC 法同时测定腰痛丸(Ⅰ)中蛇床子素、升麻素苷和橙皮苷的含量。经方法学验证,该方法简便快速、结果精确可靠,可作为腰痛丸(Ⅰ)中上述3 种指标成分的质量控制方法,为本品的质量控制标准提供有效的依据。
