基于加权基因共表达网络分析识别F2RL1为结直肠癌的核心致病基因*
2022-05-05温中庆孙雨颉赵浩楠
温中庆, 张 强, 孙雨颉, 赵浩楠, 康 宁
天津中医药大学中西医结合学院,天津 301617
结直肠癌(colorectal cancer,CRC),是发生于结直肠部位的消化道恶性肿瘤,结肠腺癌(colon adenocarcinoma,COAD)是结直肠癌中最常见的病理类型[1]。据美国癌症协会报道,2019年CRC在男性中发病率居第2位,在女性中发病率居第3位。结直肠癌患者相对5年生存率为65%,而Ⅳ期患者5年生存率仅12%[2]。早期诊断能够显著改善结直肠癌患者的预后[3],然而多数患者发现时已发展至中晚期,因此挖掘结直肠癌发生发展过程中的生物标志物具有重要意义。
随着高通量测序技术的发展,生物信息学成为了寻找生物标志物的重要手段。加权基因共表达网络分析(weighted gene co-expression network analysis,WGCNA)是一种系统生物学方法,用于检测高度相关的基因群组成的基因模块与特定的临床性状之间的相关性[4],已经被广泛应用于鉴定与多种癌症临床特征相关的基因模块,包括用于鉴定与结直肠癌复发[5]、微卫星不稳定[6]等相关的基因模块。
GEO(The Gene Expression Omnibus)是一个存储高通量基因表达数据集的公共数据库[7]。本研究采用NCBI-GEO数据库的GSE89076数据集[8],该数据集包含275例结直肠癌患者的肿瘤组织及其癌旁正常组织的基因表达谱。TCGA(The Cancer Genome Atlas)数据库是由美国国立卫生院启动的项目,旨在通过对30多种类型人类肿瘤进行大规模基因组测序,提供公开可用的癌症基因组数据,从而推动改进诊断标准、治疗方法,并最终预防癌症[9]。本文采用了来自TCGA-COAD的RNA-seq数据,其中包含473个结直肠癌样本及41个正常样本。
本研究基于生物信息学分析方法研究与结直肠癌发生发展相关的高风险致病基因及其相关通路,为结直肠癌的临床诊断及治疗提供潜在靶点。
1 材料与方法
1.1 数据下载与预处理
通过TCGA数据库(https://portal.gdc.cancer.gov/)下载结直肠癌样本和正常样本的基因表达数据,及其相应的临床数据。将Primary site选择为“colon”,Program选择为“TCGA”,Project选择为“TCGA-COAD”,Data category为“transcriptome profiling”,Data type为“Gene expression quantification”,Experimental strategy为“RNA-seq”,Workflow类型为“HTseq-counts”。用R语言处理数据,以|logFC|>1,adj.P<0.05为条件筛选差异表达基因(differentially expressed genes,DEGs)。
通过GEO数据库(https://www.ncbi.nlm.nih.gov/gds)下载芯片数据集GSE89076[8],芯片平台为Agilent-039494 SurePrint G3 Human GE v2 8x60K Microarray 039381(Feature Number version)。该微阵列数据集包含275例结直肠癌患者肿瘤组织及其癌旁正常组织的基因表达数据。根据注释文件将探针矩阵进行注释,转化为基因矩阵,删除重复探针,并以|logFC|>1,adj.P<0.05为条件筛选差异表达基因(DEGs)。
1.2 加权基因共表达网络分析
通过R语言中的WGCNA包[4]分别对TCGA-COAD和GSE89076[8]的数据构建共表达网络。首先,对样本进行聚类,删除离群样本;其次,通过筛选软阈值β,构建更符合无尺度网络特征的网络。通过设置每个模块最少基因数为50,切割高度为0.25进行模块聚类,并对相似模块进行合并,计算模块显著性(module significance,MS)。最后计算基因显著性(gene significance,GS)和模块身份(module membership,MM)。
1.3 差异共表达基因GO和KEGG富集及核心基因筛选
用R包VennDiagram[10]将WGCNA筛选出与临床特征相关性最强模块所包含的基因与差异表达基因取交集,作为差异共表达基因。随后通过R包clusterProfiler[11]对差异共表达基因进行GO和KEGG富集分析。随后采用STRING(https://www.string-db.org/)在线数据库[12],选择score>0.9预测候选核心基因之间的相互作用,构建蛋白质互作(protein-protein interaction,PPI)网络,用Cytoscape[13]对PPI网络进行可视化,并通过Cytoscape的CytoHubba插件计算每个节点的MCC(maximal clique centrality)[14]。在本研究中,MCC值最高的10个基因确定为候选核心基因。为了验证候选核心基因的预后价值,我们通过GEPIA2数据库[15]考察了候选核心基因与CRC患者总生存期及无病生存期之间的关系。以P<0.05为差异具有统计学意义,将其定义为核心基因,并对核心基因进行进一步分析。
1.4 人蛋白图谱数据库验证生存相关的核心基因蛋白质表达
人蛋白图谱数据库(The Human Protein Atlas,HPA,https://www.proteinatlas.org/)提供了基于免疫组织化学(immunohistochemistry,IHC)对蛋白质相对丰度的检测[16]。我们通过HPA数据库分析了生存相关核心基因在结直肠癌组织和正常组织中的蛋白表达情况。
1.5 单基因GSEA分析
为了确定核心基因相关信号通路,我们将TCGA数据集中的肿瘤样本根据基因表达中位数分为高表达组和低表达组,选择c2.cp.kegg.v7.2.symbols.gmt基因集作为参考基因集,使用Gene Set Enrichment Analysis(GSEA)[17](https://www.gsea-msigdb.org/gsea/index.jsp)软件v4.1.0进行分析,归一化富集分析得分(NES)是通过分析1000种排列来确定。根据先前的研究,我们按照|NES|>1、P<0.05和错误发现率(false discovery rate,FDR)<0.25为标准筛选基因集[18-19]。
1.6 细胞培养
人正常结肠上皮细胞HCoEpiC和结直肠癌细胞系HCT116、LoVo均来源于ATCC。HCoEpiC细胞在含有10%胎牛血清(BI,以色列)和1%青霉素-链霉素(Solarbio,中国)的高糖DMEM培养液(biosharp,中国)中培养;HCT116细胞和LoVo细胞在含有10%胎牛血清(ExCell,中国)和1%青霉素-链霉素(Solarbio,中国)的RPMI-1640培养液(biosharp,中国)中培养。所有细胞均置于37℃、5% CO2的培养箱中培养。
1.7 Western blot实验
用RIPA裂解细胞,12000 r/min离心10 min后吸出上清,通过BCA蛋白定量试剂盒进行蛋白定量。将各样品稀释至同一浓度,取等量样品在10%SDS-PAGE中电泳,使用F2RL1(Santa Cruz,1∶100稀释)及β-actin(中杉金桥,1∶3000稀释)一抗,置于4℃过夜孵育,加入二抗后置于室温孵育1 h,使用高敏化学发光试剂(Tanon,中国)显影。使用Image J进行光密度分析。
1.8 统计学方法
采用IBM SPSS 23.0进行统计分析,组间首先采用单因素方差分析,随后通过LSD事后检验进行多重比较,以P<0.05为差异有统计学意义。采用GraphPad Prism version 7.0进行统计作图。
2 结果
2.1 TCGA-COAD及GSE89076差异基因表达谱构建
通过R语言limma包[20]对TCGA数据库下载结直肠癌样本和正常样本及GSE89076数据集进行分析,并以|logFC|>1,P<0.05为条件筛选DEGs,并通过使用R语言ggplot2[21]绘制火山图和热图,其中红色代表表达上调的基因,蓝色代表表达下调的基因。在TCGA-COAD中共得到3481个DEGs,其中包括1208个上调基因和2273个下调基因(图1)。在GSE89076数据集中共得到3460个DEGs,其中包括1753个上调基因和1707个下调基因(图2)。
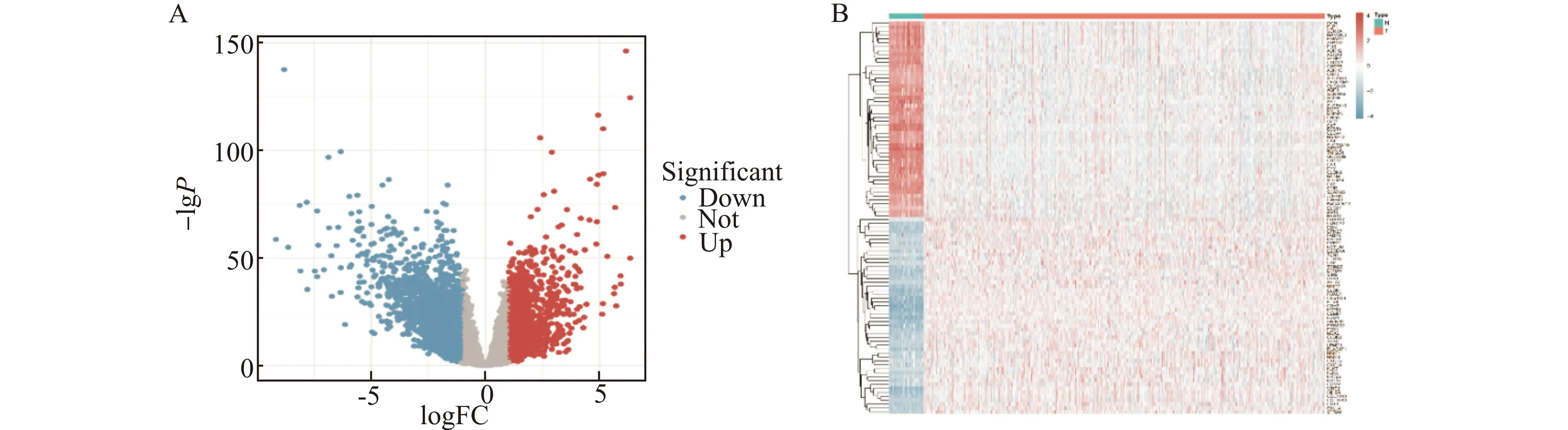
A:DEGs火山图;B:DEGs热图图1 TCGA-COAD差异表达基因(DEGs)分析Fig.1 Analysis of differentialy expressed genes(DEGs)in TCGA-COAD

A:DEGs火山图;B:DEGs热图图2 GSE89076差异表达基因(DEGs)分析Fig.2 Analysis of differentialy expressed genes(DEGs)in GSE89076
2.2 TCGA-COAD及GSE89076加权基因共表达模块构建
通过R语言的WGCNA包[4]构建加权基因共表达网络。对样本进行聚类,去除离群值后绘制样本聚类树。在TCGA-COAD中选择β=3(R2=0.9)构建无标度网络(图3);在GSE89076数据集中选择β=11(R2=0.9)构建无标度网络(图4)。本研究共确定了TCGA-COAD中的11个共表达模块及GSE89076数据集的22个共表达模块(其中包含1个无法被归类的基因组成的grey模块)。接下来绘制模块-临床特征相关性热图,以评估模块与临床特征(肿瘤与正常)之间的相关性。其中TCGA-COAD中yellow模块与肿瘤组织相关性最强(r=-0.87,P<0.05);GSE89076数据集中blue模块与肿瘤组织相关性最强(r=-0.77,P<0.05)。因此选择这2个模块为重要模块进行进一步分析。

A:样本聚类及临床特征热图;B:通过无标度拟合指数和平均连通性筛选软阈值;C:共表达网络模块聚类树状图;D:基因模块与临床特征相关性热图图3 确定TCGA-COAD中与临床特征相关的模块Fig.3 Identification of modules associated with clinical features in the TCGA-COAD

A:样本聚类及临床性状热图;B:通过无标度拟合指数和平均连通性筛选软阈值;C:基因聚类树状图,不同颜色代表不同基因模块;D:基因模块与临床特征相关性热图图4 确定GSE89076数据集中与临床特征相关的模块Fig.4 Identification of modules associated with clinical features in the GSE89076
2.3 差异共表达基因鉴定及其富集分析
将TCGA-COAD中yellow模块,GSE89076数据集中blue模块,TCGA-COAD的DEGs和GSE89076数据集的DEGs取交集,识别到436个重叠基因作为差异共表达基因(图5A)。为了进一步探究436个差异共表达基因的潜在功能,我们使用clusterProfiler包[11]对其进行了富集分析。KEGG富集分析主要富集在补体系统、细胞因子-细胞因子受体的相互作用途径、肿瘤坏死因子信号通路、细胞粘附分子途径和谷胱甘肽代谢途径等信号通路(图5B)。通过GO富集分析,其生物学过程(biological process,BP)主要在脂质分解和脂肪酸代谢过程中富集;细胞成分(cellular component,CC)分析主要涉及细胞顶端和顶端质膜;分子功能(molecular function,MF)分析中主要富集在阴离子跨膜转运体活性和磷酸酯水解酶活性(图5C)。

A:DEGs与重要共表达模块之间的韦恩图,共436个交集基因作为差异共表达基因;B:差异共表达基因的KEGG富集分析;C:差异共表达基因的GO富集分析图5 确定差异共表达基因及其GO、KEGG富集分析Fig.5 Identification of differentially co-expressed genes and their results from GO and KEGG analysis
2.4 候选核心基因筛选
随后采用STRING数据库[12]构建436个基因的蛋白质-蛋白质互作(PPI)网络,通过Cytoscape[13]对网络进行可视化,并采用CytoHubba插件的MCC算法从PPI网络中筛选排名前10的基因作为候选核心基因[14]:BDKRB2、GCG、EDN2、EDN3、P2RY2、P2RY1、F2RL1、GNA11、SST、ADRA2(图6)。
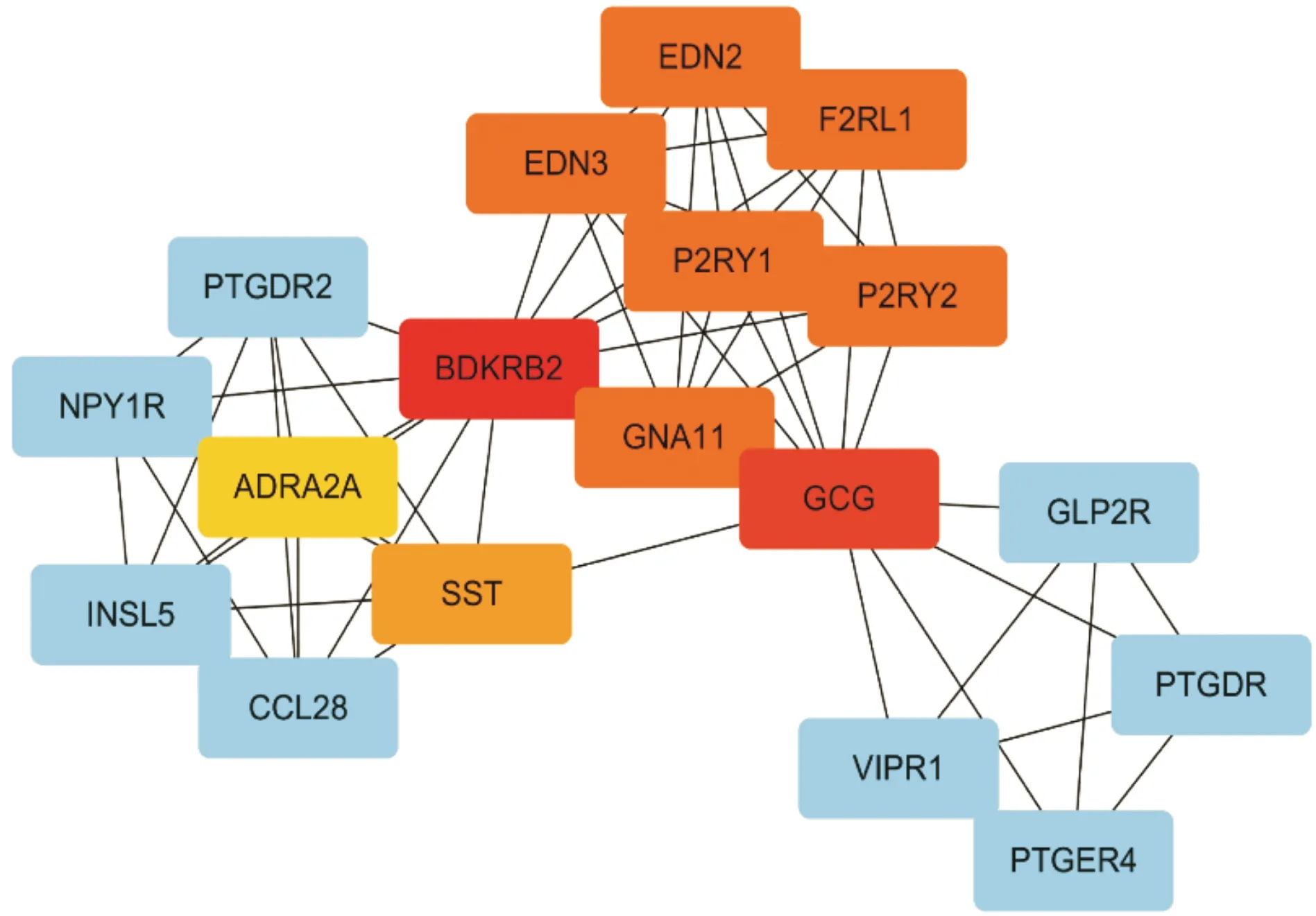
红色节点代表MCC得分较高的基因,黄色节点代表MCC得分较低的基因图6 PPI网络及候选核心基因可视化Fig.6 Visualization of the PPI network and the candidate hub genes
2.5 核心基因筛选及其mRNA和蛋白质表达模式验证
我们用GEPIA2数据库[15]考察了10个候选核心基因的预后价值。Kaplan-Meier分析显示仅F2RL1的低表达与较差的总生存期(overall survival,OS)相关(图7),而低表达F2RL1与无病生存期(disease-free survival,DFS)无显著相关性(图8)。随后,通过GEPIA2数据库[15],对F2RL1在人体正常组织样本和COAD样本中的mRNA表达差异进行分析,发现F2RL1的mRNA水平在肿瘤组织中显著降低(图9)。此外,分析HPA数据库[16],显示CRC肿瘤组织中F2RL1的蛋白质水平低于正常组织(图10)。上述研究证实,F2RL1在CRC组织中表达下调,且F2RL1的低表达与CRC患者总生存期较短相关。

A:ADRA2;B:BDKRB2;C:EDN2;D:EDN3;E:F2RL1;F:GCG;G:GNA11;H:P2RY1;I:P2RY2;J:SST图7 通过GEPIA2在线工具分析10个候选核心基因与COAD患者总生存期的关系Fig.7 Analysis of relationship between 10 candidate hub genes and OS of COAD patients by using the GEPIA2 online tool
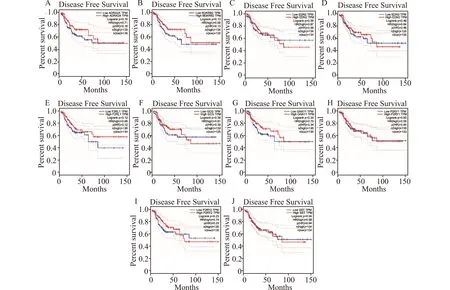
A:ADRA2;B:BDKRB2;C:EDN2;D:EDN3;E:F2RL1;F:GCG;G:GNA11;H:P2RY1;I:P2RY2;J:SST图8 通过GEPIA2在线工具分析10个候选基因与COAD患者无病生存期的关系Fig.8 Analysis of relationship between 10 candidate hub genes and DFS of COAD patients by using the GEPIA2 online tool

*P<0.05图9 使用GEPIA2对TCGA数据库的275个COAD样本和41个正常样本中F2RL1 mRNA水平进行差异表达分析Fig.9 Analysis of differential expression of F2RL1 mRNA in 275 COAD samples and 41 normal samples in the TCGA database by using GEPIA2
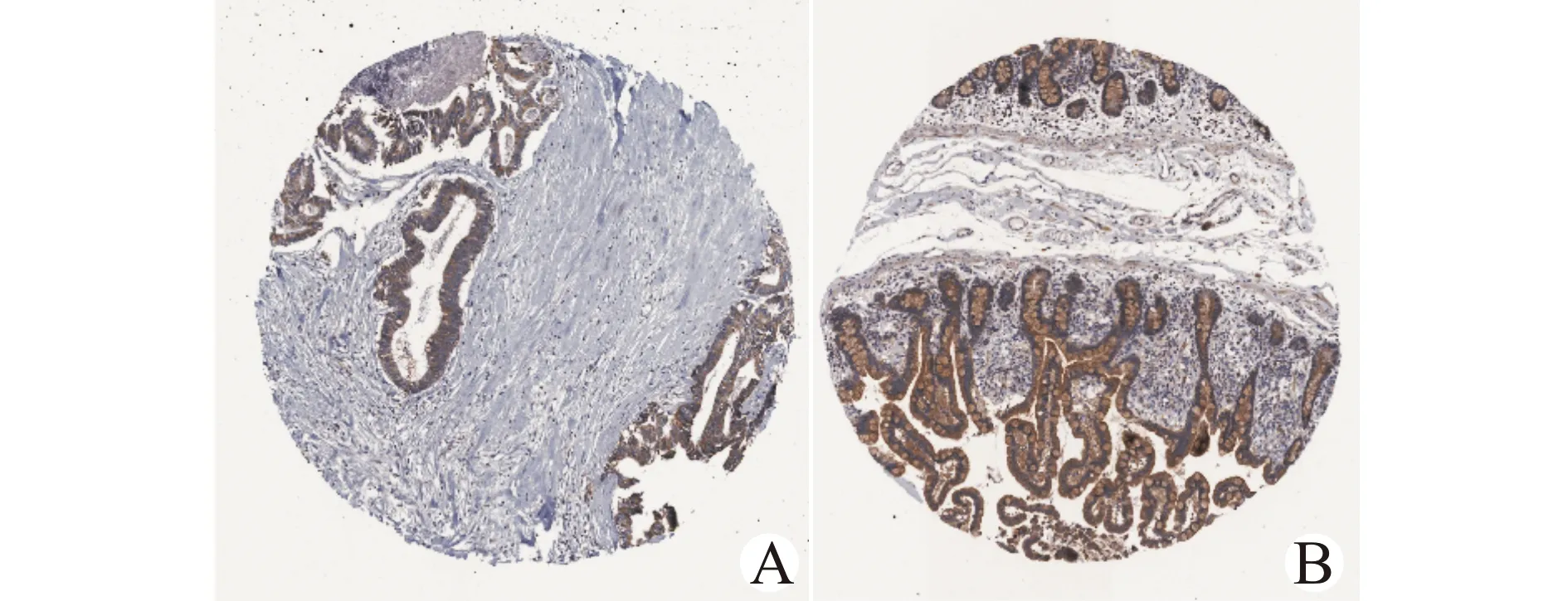
A:F2RL1在肿瘤样本中的表达;B:F2RL1在正常样本中的表达图10 HPA数据库中免疫组织化学染色显示F2RL1蛋白在CRC组织和正常组织中的表达(×100)Fig.10 Immunohistochemistry analysis of F2RL1 in CRC tissues and normal tissues from HPA database(×100)
2.6 对F2RL1进行单基因GSEA分析
为了进一步确定核心基因的潜在功能,本研究对TCGA-COAD来源的数据进行了基于KEGG基因集的单基因GSEA。单基因GSEA揭示了F2RL1的上调与核苷酸切除修复信号通路(NES=1.77,P<0.01)、P53信号通路(NES=1.70,P=0.009)、蛋白酶体信号通路(NES=1.60,P=0.034)、错配修复信号通路(NES=1.57,P=0.037)等呈正相关(图11)。
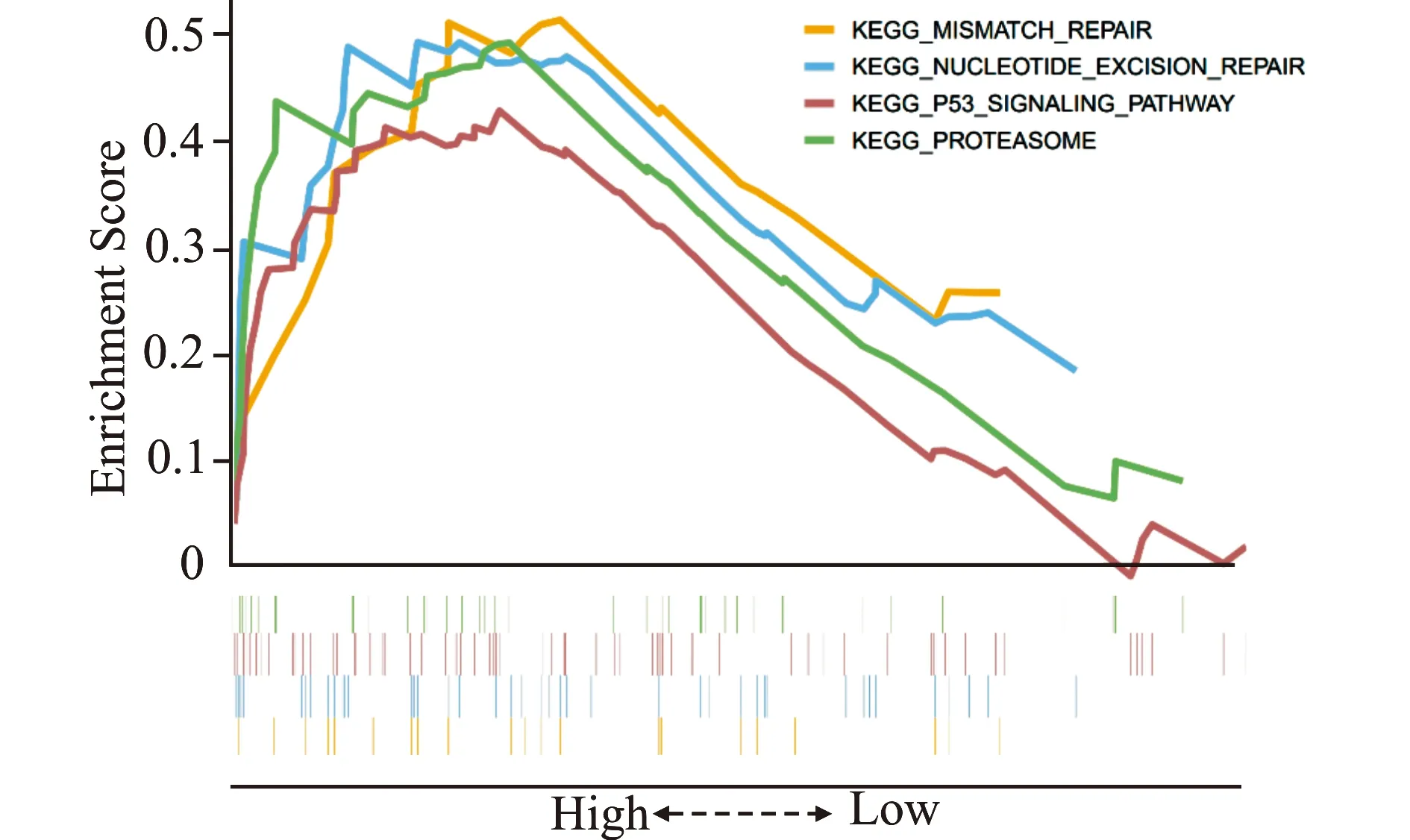
图11 基于TCGA-COAD数据对F2RL1进行单基因GSEA分析(仅列出5个最具有代表性的常见KEGG通路)Fig.11 Single-gene GSEA analysis of F2RL1 based on TCGA-COAD data(only top five representative KEGG pathways are listed)
2.7 F2RL1在人结直肠细胞系中的表达
通过Western blot验证了F2RL1在人正常结肠上皮细胞HCoEpiC和结直肠癌细胞HCT116、LoVo中的表达水平。与生信分析预测结果一致,结直肠癌细胞中F2RL1的蛋白表达较正常结肠上皮细胞中的蛋白表达显著下调(图12)。

与HCoEpiC比较,*P<0.05图12 F2RL1蛋白质表达水平验证Fig.12 Verification of expression levels of F2RL1 protein
3 讨论
CRC是最常见的胃肠道肿瘤类型[22],在中国的发病率呈上升趋势[23]。超过90%的CRC由结肠息肉发展而来[24],如果在息肉阶段治愈,防止癌变,5年生存率可达90%以上[25],但是晚期患者的5年生存率<10%。目前,结肠镜和组织活检是主要用于早期发现CRC的手段,然而结肠镜是一种侵入性手术,成本高,且需要较高水平的专业技术。因此,需要寻找更好的CRC特异性预后和进展生物标志物及治疗靶点。在本研究中,我们使用WGCNA和差异表达基因分析,在TCGA-COAD和GSE89076数据集中鉴定了436个表达趋势相同的差异共表达基因。进一步通过PPI网络确定了BDKRB2、GCG、EDN2、EDN3、P2RY2、P2RY1、F2RL1、GNA11、SST、ADRA2这10个候选核心基因。将10个候选基因进行预后价值验证,最终确定F2RL1为核心基因。
F2RL1(F2R like trypsin receptor 1)编码的蛋白为蛋白酶激活受体2(proteinase-activated receptor 2,PAR2),属于G蛋白偶联受体家族[26]。在以往的研究中发现F2RL1与炎症[27]、疼痛[28]、胆固醇与脂质代谢[29]、肥胖[30]、血管生成等相关。除此之外,据文献报道,F2RL1的mRNA和蛋白表达在炎症性肠病和结直肠癌样本中均下调,且F2RL1下调与较短的生存期相关[31]。有趣的是,Kawaguchi等[32]发现F2RL1促进肠道肿瘤的发生;也有研究表明在肺腺癌中低表达F2RL1与较好的预后相关[33];在肝细胞癌中F2RL1促进迁移和侵袭[34]。本研究通过对F2RL1进行单基因GSEA分析,确定了核苷酸切除修复信号通路、P53信号通路、蛋白酶体信号通路、错配修复信号通路在高表达F2RL1组中被上调。核苷酸切除修复(NER)是保护细胞免受基因组损伤的主要DNA修复途径之一,这条通路的中断会促进癌症的发展[35],并且癌细胞在参与DNA修复的相关基因中显示出各种类型的突变和异常表达。这些改变会导致基因组不稳定并促进癌症发生和癌症进展过程。因此DNA修复中的这些缺陷也被认为是癌症治疗的合适靶点[36];转录因子P53主要通过调控其下游凋亡、细胞周期、DNA修复、衰老等相关靶基因转录,在抑制肿瘤中发挥极其重要的作用,其突变或失活是几乎所有肿瘤的标志[37-38];泛素蛋白酶体通过调控细胞中大多数蛋白质的降解,从而参与许多细胞过程的调控[39],其功能障碍与多种肿瘤的发生相关[40];错配修复系统是维持DNA稳定性的保证[41],DNA错配修复缺陷会导致高突变表型,从而导致肿瘤的发生[42]。这些结果进一步证实了F2RL1可能在CRC发生发展中通过上述通路发挥重要作用。
综上所述,通过WGCNA和差异表达基因分析,我们的研究确定了CRC中具有预后价值的核心基因F2RL1,并且进一步验证了F2RL1在人结直肠癌细胞系中表达下调。虽然我们通过对多个数据库的CRC相关数据进行了生物信息学分析,但识别核心基因只是筛选CRC生物标志物及治疗靶点的第一步,我们仍需要通过一系列体外体内的实验验证F2RL1在CRC发生发展和预后中的价值,以及其发挥作用的分子机制。
