基于茚并咔唑与均三嗪的N 型主体材料的合成及其在绿色磷光器件中的性能研究
2022-04-29檀笑风肖文平霍延平
刘 宇,檀笑风,陈 迁*,戴 雷*,肖文平,霍延平
(1. 广东工业大学 轻工化工学院,广东 广州 510006;2. 广东阿格蕾雅光电材料有限公司,广东 佛山 528300)
1 引 言
有机电致发光二极管(OLED)被认为是新一代显示和照明器件,具有效率高、重量轻、制造面积大、机械灵活性好、加工成本低等优点[1],在过去的20 年里,有机显示技术得到了迅猛的发展。在磷光有机电致发光二极管(PhOLED)里,因有过渡金属的参与,单线态激子可通过快速系间窜越(ISC)转化为三线态激子,然后所有三线态激子以磷光的形式辐射衰减,从而捕获所有单线态和三线态激子,实现理论上100%的内部量子效率(IQE)[2-3]。但传统磷光材料大多存在三重态-三重态激子淬灭(TTA)效应,降低了器件性能,为了解决这个问题,通常将磷光材料均匀地分散在主体材料中。因此,开发高性能的主体材料,对于提高器件的整体性能具有重要意义[4]。
高效电致发光器件的主客体系统通常需要满足以下要求:(1)具有良好的热稳定性和成膜能力,以延长器件的使用寿命[5-6];(2)主体材料的三重态能级(Et)应高于掺杂材料的三线态能级,从而实现从主体到客体的完全能量传递,并将三线态激子限制在发光层内[7-9];(3)主体的最低未占据分子轨道(LUMO)能级和最高占据分子轨道(HOMO)能级应与相邻层的LUMO 能级和HOMO 能级相匹配,以便于在较低的驱动电压下进行电荷注入[10-11];(4)主体材料应具有双极电荷传输特性,以扩宽发光层内的电荷复合区,从而提高器件效率[12-13]。然而,单个主体通常很难满足以上所有要求,在这方面,一个较为成熟的策略是通过至少两种主体材料的混合来构建器件的发光层[14]。两种主体材料的混合,特别是N 型主体与P 型主体组合,同时保证了发光层中优越的电子传输能力和空穴传输能力,赋予了双极传输的特性,拓宽了激子复合的区域,并平衡了载流子的注入和传输,可以显著提高器件性能[15]。很多基于N 型主体与P 型主体的组合往往能形成激基复合物。在激基复合型混合主体的开发中,需要将P 型主体的HOMO 与N 型主体的LUMO 进行有效匹配[16]。此外,两种主体材料都需要有较高的三重态能量,以避免形成的激基复合物的三重态能量过低,导致三重态激子从客体材料向激基复合物的转移[17]。1,3,5-三嗪(TRz)由于其高度的缺电子构型,具有良好的电子传输能力,茚并[2,1-b]咔唑由于其良好的富电子构型,具有良好的空穴传输能力,这两种结构均被广泛应用于给体-受体(D-A)发光体系的设计[18-20]。
本文在1,3,5-三嗪(TRz)和7,7-二甲基-5,7-二氢茚并[2,1-b]咔唑的基础之上,设计合成了一系列具有较深LUMO 能级的主体材料,使用密度泛函理论(DFT)建立了结构与其性能之间关系,并对其热力学性质、电化学性能和光物理性质进行了研究。最后与P 型主体9,9'-二([1,1'-联苯]-4-基)-3,3'-联-9H-咔唑(NBPBC)进行1∶1 混合,搭配磷光客体Pt(BPP),制备了混合型主体的绿光器件(PhOLED)。所有器件均实现了低启动电压(2.1~2.3 V),并随着N 型主体LUMO 能级的加深,器件寿命也不断增加,其中DCNTRz∶NBPBC(器件III)在4 000 cd·A-2拥有282 h 的最长寿命(LT95),比DTRz∶NBPBC(器件I)的寿命提升了54%,同时还具有45.7 lm·W-1的功率效率、46.5 cd·A-1的电流效率以及14.3%的外量子效率。
2 实 验
2.1 中间体和主体的合成
三个主体分子合成路线如图1 所示,中间体2b,选用醋酸钯为催化剂,以K4Fe(CN)6·3H2O为氰源,由2a 通过芳基卤化物的氢化反应得到。中间体3c,选用二氯双[二叔丁基-(4-二甲基氨基苯基)膦]钯(II)为催化剂,在叔丁醇钠的作用下,由3a 和3b 通过Buchwald-Hartwig 偶联反应得到。3 个主体目标产物,都直接或通过中间体由Buchwald-Hartwig 偶联反应得到,其中DBTRz用到了一锅法合成。合成的中间产物和目标产物的结构已通过质谱和核磁共振谱进行了确认。

图1 化合物DTRz、DCNTRz、DBTRz 的合成。Fig.1 Synthetic routes of compound DTRz,DCNTRz and DBTRz.
2.2 材料合成
DTRz 的合成过程如下:取500 mL 烧瓶,投入(3-溴 苯 基)-4,6-二 苯 基-1,3,5-三 嗪(1a)(4.10 g,10.6 mmol,1.0 eq)、7,7-二甲基-5,7-二氢茚并[2,1-b]咔唑(1b)(3.0 g,10.6 mmol,1.0 eq)、醋酸钯(118.2 mg,0.53 mmol,0.05 eq)、三叔丁基磷(214 mg,1.06 mmol,0.1 eq)、叔丁醇钠(2.54 g,26.5 mmol,2.5 eq)、甲苯(100 mL);在氮气保护下,90oC 反应8 h,冷却至室温,加入正己烷析出固体后抽滤,并用甲醇洗涤,取固体,用四氢呋喃溶解后,快速粗硅胶过滤除去钯催化剂,溶剂在减压下馏去,随后将固体用四氢呋喃/甲醇重结晶得到黄色固体2.01 g,产率为67%,升华后,HPLC 纯度为99.93%。1H NMR(400 MHz,CDCl3)δ:9.05(s,1H),8.95~8.86(m,1H),8.83~8.69(m,4H),8.50(s,1H),8.25(d,J=7.6 Hz,1H),7.96~7.81(m,3H),7.67~7.47(m,8H),7.47~7.26(m,6H),1.54(s,6H)。13C NMR(101 MHz,CDCl3)δ:171.86,171.00,153.36, 153.31, 141.27, 141.16, 139.67,138.52, 138.38, 135.94, 132.70, 132.56,130.95, 130.33, 129.00, 128.67, 127.85,127.57, 127.08, 126.34, 125.80, 123.74,123.09, 122.56, 120.22, 120.18, 119.39,111.40,109.78,103.99,46.81,27.95。 MS(MALDI-TOF)[m/z]: calcd for C42H30N4,590.25;found:613.30[M+Na]+。
2-(3-氰基-5-氯苯基)-4,6-二苯基-1,3,5 三嗪(2b)的合成:在1 000 mL 单口瓶中加入2-(3-溴-5-氯苯基)-4,6-二苯基-1,3,5 三嗪(2a)(10.0 g,23.6 mmol,1.0 eq)、K4Fe(CN)6·3H2O(8.0 g,23.6 mmol,0.8 eq)、四(三苯基膦)钯(1.36 g,1.18 mmol,0.05 eq)、N,N- 二 甲 基 甲 酰 胺(340 mL);随后在氮气保护下,150 ℃回流8 h,冷却至室温,往反应液里加水后抽滤后取固体烘干,粗产物用二氯甲烷/正己烷(1∶3)柱层析,溶剂在减压下馏去,得白色固体6.0 g,产率70%,HPLC 纯 度99.51%。1H NMR(400 MHz,CDCl3)δ:8.90(s,2H),8.74~8.69(m,4H),7.83(t,J=1.7 Hz,1H),7.62(dt,J=24.0,7.1 Hz,6H)。
DCNTRz 的合成:取500 mL 单口瓶两个,分别等量投入2-(3-氰基-5-氯苯基)-4,6-二苯基-1,3,5 三嗪(2b)(3.9 g,10.6 mmol,1.0 eq)、7,7-二甲基-5,7-二氢茚并[2,1-b]咔唑(2c)(3.0 g,10.6 mmol,1.0 eq)、三(二亚苄基茚丙酮)二钯(0)(0.49 g,0.53 mmol,0.05 eq)、2-二 环 己 基膦-2',4',6'-三 异 丙 基 联 苯(1.0 g,2.12 mmol,0.2 eq)、碳酸铯(8.6 g,26.5 mmol,2.5 eq)、二甲苯(110 mL);在氮气保护下,120oC 回流9~10 h,冷却至室温,合并上面各反应液,加入过量正己烷析出固体后抽滤,将固体用甲醇洗涤,取固体,用四氢呋喃溶解后,快速用粗硅胶过滤除去钯催化剂,溶剂在减压下馏去,随后将固体用二氯甲烷/甲醇重结晶,得到黄色固体5.90 g,产率71%。 升 华 后,HPLC 纯 度 为99.93%1H NMR(400 MHz,CDCl3)δ:9.29(s,1H),9.15(s,1H),8.75(d,J= 7.1 Hz,4H),8.48(s,1H),8.25(d,J= 7.7 Hz,1H),8.16(s,1H),7.89(d,J= 7.4 Hz,1H),7.62(t,J= 7.3 Hz,2H),7.58~7.52(m,5H),7.43(ddd,J= 22.7,13.7,7.1 Hz,5H),7.32(t,J= 7.4 Hz,1H),1.55(s,6H).13C NMR(101 MHz,CDCl3)δ:172.42,170.68, 169.39, 168.59, 153.84, 149.40,140.20, 139.80, 138.79, 135.63, 134.79,134.17, 133.39, 132.56, 131.97, 130.11,129.88, 129.80, 129.03, 128.50, 128.27,128.20, 126.06, 124.27, 123.68, 122.56,122.12, 120.80, 120.51, 118.54, 118.10,117.21, 115.01, 112.68, 112.56, 111.10,109.79,104.60,103.02,48.95,47.11,43.66,28.78,26.89。MS(MALDI-TOF)[m/z]:calcd for C43H29N5,615.24;found:616.30[M+H]+。
5-(3-氯-5-(4,6-二苯基-1,3,5-三嗪-2-基)苯基)-7,7-二甲基-5,7-二氢茚并[2,1-b]咔唑(3c)的合成:取500 mL 单口瓶两个,投入7,7-二甲基-5,7-二 氢 茚 并[2,1-b]咔 唑(3b)(6.67 g,23.65 mmol,1.0 eq)、2-(3-溴-5-氯苯基)-4,6-二苯基-1,3,5 三嗪(3a)(10.0 g,23.65 mmol,1.0 eq)、二氯双[二叔丁基-(4-二甲基氨基苯基)膦]钯(II)(0.592 g,1.18 mmol,0.02 eq)、叔丁醇钠(6.82 g,71.0 mmol,3.0 eq)和二甲苯(200 mL),在氮气保护下,100oC 反应16 h,反应液冷却至室温,将反应液缓慢滴加至500 mL 甲醇中,析出黄色固体,抽滤取固体,用二氯甲烷溶解,快速粗硅胶过滤除去钯催化剂,溶剂在减压下馏去,粗产物用二氯甲烷/正己烷重结晶后得到淡黄色粉末20.0 g,产率67%,HPLC 纯度99.6%。1H NMR(400 MHz,CDCl3)δ:8.96(s,1H),8.86(s,1H),8.76(d,J= 7.3 Hz,4H),8.49(s,1H),8.24(d,J= 7.8 Hz,1H),7.89(d,J= 7.3 Hz,2H),7.61(t,J=7.2 Hz,2H),7.54(dd,J= 14.0,7.2 Hz,6H),7.49~7.43(m,2H),7.43~7.34(m,2H),7.31(t,J= 7.3 Hz,1H),1.55(s,6H)。
DBTRz 的合成:取250 mL 单口瓶两个,首先各投入5-(3-氯-5-(4,6-二苯基-1,3,5-三嗪-2-基)苯基)-7,7-二甲基-5,7-二氢茚并[2,1-b]咔唑(3c)(5.0 g,8.0 mmol,1.0 eq)、联硼酸频那醇酯(4.0 g,16.0 mmol,2.0 eq)、Pd2(dba)3(160 mg,0.16 mmol,0.02 eq)、三 环 己 基 膦 四 氟 硼 酸 盐(117 mg,0.32 mmol,0.04 eq)和DMF(100 mL),氮气保护,80oC 反应16 h,原料5-(3-氯-5-(4,6-二苯基-1,3,5-三嗪-2-基)苯基)-7,7-二甲基-5,7-二氢茚并[2,1-b]咔唑(3c)反应完全后,各加入2-氯-4,6-二苯基-1,3,5-三嗪(3d)(4.25 g,16 mmol,2.0 eq),二氯双[二叔丁基-(4-二甲基氨基苯基)膦]钯(II)(117 mg,0.16 mmol,0.02 eq),碳酸钾(3.3 g,24 mmol,3.0 eq)和水30 mL,在氮气保护下,90oC 反应24 h 后反应结束。将反应液中加入一倍体积的甲醇,搅拌5 min 左右,抽滤取固样,烘干,用邻二氯苯(o-DCB)溶解,用粗硅胶过滤除去钯催化剂,在溶液中加入一倍体积的正己烷,常温搅拌重结晶4 h 左右,抽滤,烘干,得到黄色固体。后续再用二氯甲烷80 mL ,50oC 打浆4 h,抽滤,烘干。得到黄色粉末6.0 g,产率50%,HPLC 纯 度99.08%。1H NMR(400 MHz,CDCl3)δ:10.32(s,1H),9.27(d,J= 1.4 Hz,2H),8.86(d,J= 6.9 Hz,8H),8.55(s,1H),8.30(d,J= 7.6 Hz,1H),7.92(d,J= 7.0 Hz,1H),7.68(s,1H),7.65~7.57(m,13H),7.48(t,J= 7.2 Hz,1H),7.43(t,J= 6.9 Hz,2H),7.40~7.38(m,1H),7.32(d,J= 6.9 Hz,1H),1.56(s,6H).13C NMR(101 MHz,CDCl3)δ:163.31, 152.85, 152.25, 151.72, 150.48,149.48, 148.12, 145.75, 141.61, 139.85,138.71, 133.23, 130.42, 128.89, 127.66,126.80, 123.95, 122.82, 122.57, 121.69,121.19, 121.09, 120.99, 120.92, 120.61,119.59,117.92,114.39,32.41,31.20。 MS(MALDI-TOF)[m/z] : calcd for C57H39N7,821.60;found,839.30[M+H2O]+。
2.3 器件制备与表征
本文使用的氧化铟锡(ITO)基板厚度为0.7 mm,其中ITO 的厚度为110 nm(方阻约15 Ω/□),蒸镀前的玻璃基板经过去离子水、清洗剂、四氢呋喃、二氯甲烷在超声波清仪中分别清洗20 min,最后用氮气吹干。ITO 表面经过基于电容耦合的氧等离子处理装置,进行表面氧等离子处理15 min。器件的发光面积为0.09 cm2。OLED 器件的制备在有机热蒸发体系中进行,在5×10-4Pa 的真空环境下进行蒸镀,有机材料的蒸发速率控制在0.03 nm/s,LiQ 的蒸镀速率为0.02 nm/s,Ag 的蒸镀速率控制在0.1 nm/s。利用石英晶体震荡片来检测薄膜厚度。随后器件的光电特征使用Keithley 2400 Source Meter 和PR655 光谱辐射计进行测量,获得器件电流密度、效率、亮度和电致发光光谱。单载流子器件的阻抗谱是通过Soulartron1260 阻抗分析仪进行测量,在样品上施加0.1 V 的正弦信号电压,频率范围为1~108 Hz。
3 结果与讨论
3.1 理论模拟
主体化合物的前线轨道(HOMO 和LUMO)对设计磷光主体材料具有非常重要的意义。为了从理论上探索分子结构与材料性能的关系,获得化合物的电子结构分布,采用密度泛函理论(DFT)中的B3LYP 方法,结合6-31G*基组,计算优 化 了DTRz、DBTRz 和DCNTRz 在 基 态 的 几何构型及前线轨道分布,全部计算工作采用Gaussian 09 软件完成。
图2 中显示了DTRz、DBTRz 和DCNTRz 三个分子优化后的基态几何及其前线轨道分布。3 个分子的HOMO 主要离域在以茚并咔唑为中心的富电子单元上,这与其供电子基团的身份是一致的。3 个分子的LUMO 则都分布在以二苯基三嗪和氰基为主的区域。对比DTRz,DBTRz比其多一个二苯基三嗪电子受体,DCNTRz 比其多一个氰基电子受体,因而后两者的LUMO 分布区域更分散一些。强吸电子基的引入通常会导致分子轨道简并后的LUMO 能级加深。由理论 计 算 得 出 的DTRz、DBTRz 和DCNTRz 的LUMO 分别是-2.24,-2.39,-2.57 eV,与其吸电子能力的增强方向一致,也与后续的实验结果相一致。3 个分子计算得出的HOMO 能级分别为-5.35,-5.36,-5.56 eV,DCNTRz 相对更深的HOMO 应该是由于氰基的强吸电子作用将HOMO 轨道推离了与氰基相连的苯环而导致的,此结果同样与后续的实验数据相吻合。

图2 化合物的HOMO 和LUMO 能级分布图Fig.2 HOMO and LUMO distribution of compounds
3.2 热力学性质
高效OLED 器件的主体材料要求具有较高的热稳定性,以防止蒸镀时出现热分解,或高温下出现玻璃化转变从而造成期间寿命和效率的损失。在此用差示扫描量热法(DSC)和热重分析法(TGA)研究了DTRz、DBTRz 和DCNTRz的玻璃化转变温度(Tg)和分解温度(Td,相当于5% 的 失 重)。如 图3 所 示,DTRz、DBTRz 和DCNTRz 的玻璃化转变温度分别是139.8,177.3,161.9 ℃,热 分 解 温 度 分 别 为411.4,499.8,419.1 ℃。所有的材料都表现出极好的热稳定性,符合器件制备的热物理要求。这其中,DBTRz 由于其相对更大的分子量,具有更大的玻璃化转变温度和热分解温度。

图3 化合物DTRz、DBTRz 和DCNTRz 的TGA 和DSC曲线。Fig.3 TGA&DSC curves of DTRz,DBTRz and DCNTRz.
3.3 电化学性质
循环伏安法测试可以得到材料的氧化还原电位,分别近似对应着材料的垂直电离能(VIP)和垂直电子亲和能(VEA),这两个能量通常与材料的HOMO 和LUMO 存在着一定的正相关性。因此,我们通过循环伏安法来定性分析材料的前线 轨 道。 如 图4,化 合 物DTRz、DBTRz 和DCNTRz 分别在1.26,1.20,1.32 V 出现第一个氧化峰,计算其HOMO 能级分别为-5.50,-5.46,-5.59 eV。在-1.28,-1.15,-1.11 V出现第一个还原峰,计算其LUMO 分别为-2.95,-3.11,-3.16 eV。由此得出电化学基础带隙分别为,2.55,2.35,2.43 eV(计算公式见表1 附注),测试结果与理论计算结果(3.11,2.97,2.99 eV)高度一致。可见分子中额外引入的二苯基三嗪和氰基官能团确实会由于其强大的吸电子能力,拉深分子的LUMO 轨道能量,而这正是我们提升器件性能的关键所在。

图4 化合物DTRz、DBTRz 和DCNTRz 在N,N-二甲基甲酰胺溶液中的循环伏安曲线图。Fig.4 Cyclic voltammetry curves of DTRz,DBTRz,and DCNTRz in DMF solution.
3.4 光物理性质
为 了 研 究DTRz、DBTRz 和DCNTRz 的 光物理性质,在室温下记录了DTRz、DBTRz 和DCNTRz 在甲苯溶液中的紫外/可见吸收光谱和光致发光光谱。如图5 和表1,对于不含金属的共轭分子,发光通常来自π-π*跃迁、n-π*跃迁。图 中 显 示DTRz、DBTRz 和DCNTRz 在290 nm左右处的吸收峰主要来自茚并[2,1-B]咔唑单元的π-π*吸收与n-π*吸收。在300~400 nm 的吸收具有明显的位于给电子基团和吸电子基团之间的内电荷转移(ICT)特征。3 个材料末端吸收均在400 nm 附近,其光学带隙(Eoptg)则可通过吸收末端计算(Eoptg=1 240/λoffset)得出,分别为2.96,2.85,2.87 eV。DTRz、DBTRz 和DCNTRz 在甲苯溶液中的最大荧光发射峰分别位于462,483,498 nm,半峰宽分别为61,59,73 nm。与DTRz相比,DBTRz 和DCNTRz 分别出现了不同程度的红移,这与其引入额外的吸电子基团,导致分子轨道简并后的前线轨道能量降低有关。主体材料的三重态能级对于PhOLED 而言是至关重要的。如果主体材料的三重态能级不能显著高于客体材料,容易出现从客体材料三重态到主体三重态的逆向能量传递,从而极大地降低器件效率。为 了 研 究DTRz、DBTRz 和DCNTRz 的 三重态能级,在77 K 的温度下测量了薄膜状态下的磷光发射光谱。其三重态能级可由低温磷光发射峰起始边计算得出,所有化合物具有相对接近的三重能级(2.71~2.83 eV)。由于这3 个材料的三重态能量均高于后续选择的客体材料Pt(BPP)(Et=2.49 eV),因此足以将三重态激子限制在客体掺杂剂上,并阻止这部分能量从客体掺杂剂向主体材料的回传。
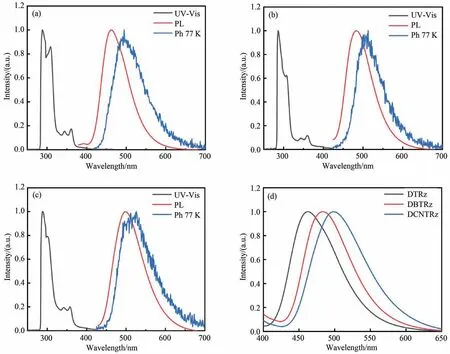
图5 (a)DTRz、(b)DBTRz 和(c)DCNTRz 在甲苯溶液中的紫外-可见光吸收、荧光和薄膜状态下的低温(77 K)磷光光谱;(d)在甲苯溶液中DTRz、DBTRz 和DCNTRz 的PL 光谱。Fig.5 UV-Vis absorption,fluorescence(in toluene solution)and phosphorescence(at 77 K in thin film)spectra of(a)DTRz,(b)DBTRz,(c)DCNTRz;(d)PL spectra of DTRz,DBTRz,DCNTRz in toluene solution.

表1 DTRz、DBTRz 和DCNTRz 的光物理、热稳定性和电化学性质Tab. 1 Photophysical,thermal stability and electrochemical properties of DTRz,DBTRz and DCNTRz
对 于DTRz、DBTRz 和DCNTRz 三 种 拥 有较深的LUMO 能级的N 型主体材料,需要从高三重态能级的主体中选择一种具有合适HOMO能级的主体材料。9,9'-二([1,1'-联苯]-4-基)-3,3'-联-9H-咔唑(NBPBC)作为空穴传输型(P 型)主体,其HOMO 为-5.38 eV,比DTRz、DBTRz和DCNTRz 的HOMO 能级略微偏浅,LUMO 能级 为-1.97 eV,比DTRz、DBTRz 和DCNTRz的LUMO 能级明显更浅,能很好地与DTRz、DBTRz 和DCNTRz 相匹配。在此基础上得到了3 种混合主体,NBPBC∶DTRz、NBPBC∶DBTRz和NBPBC∶DCNTRz。为了研究这种混合主体的光学性质,将其按照质量比1∶1 进行混合,并与单一材料的薄膜进行了对比。混合主体是否能形成激基复合物,可以通过固态混合膜的荧光光谱的红移来判断。如果能形成激基复合物,则激基复合物的HOMO 应该为P 型材料的HOMO,激基复合物的LUMO 应该为N 型材料的LUMO。因此,混合主体相对于N 型主体的偏红程度,实 际 上 取 决 于NBPBC 的HOMO 与3 种N 型主体材料的HOMO 的差值。从图6 中可以看出3 种混合物的光谱均比各自的单一分子光谱要略微偏红一些,特别是DBTRz 和DCNTRz。DTRz 的混合主体没有出现明显偏红,这同样是由于其LUMO 能级与P 型主体的LUMO 能级过于接近导致的。各混合主体发射峰均与磷光掺杂剂Pt(BPP)的吸收有不同程度的重叠,从而能顺利实现从主体到掺杂剂的Förster 能量传递。
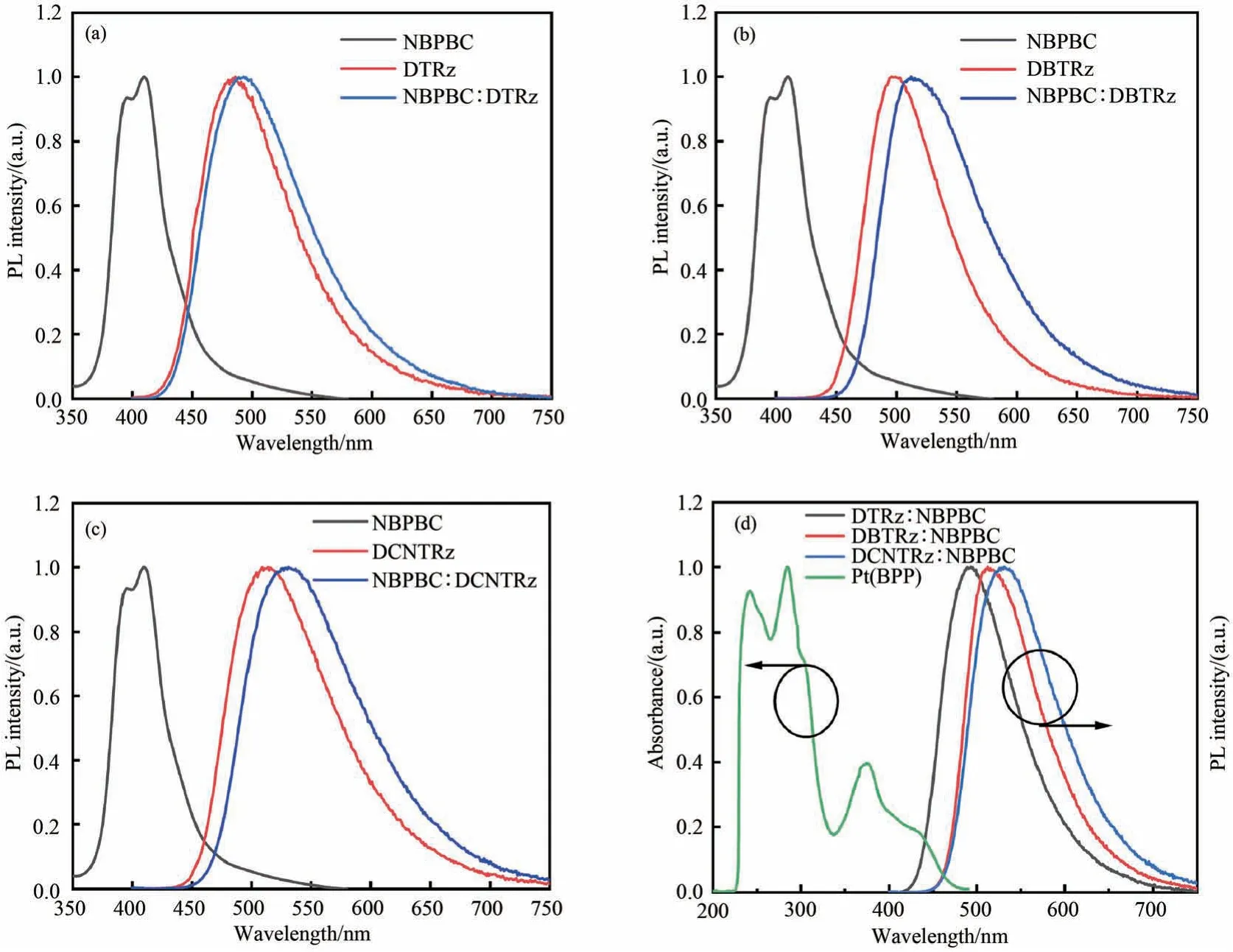
图6 (a)~(c)各主体的荧光光谱与(d)混合型主体的荧光光谱与磷光客体Pt(BPP)的吸收Fig.6 PL emission spectra of each host and mixed hosts. (a)NBPBC∶DTRz;(b)NBPBC∶DBTRz;(c)NBPBC∶DCNTRz;(d)Mixed hosts’ PL of DTRz,DBTRz,DCNTRz and UV-Vis of Pt(BPP).
3.5 单载流子测试
为了进一步研究DTRz、DBTRz 和DCNTRz的电子传输能力,制备了单电子(EOD)器件,EOD 器件结构为Ag(100 nm)/BCP(10 nm)/Ntype Host(40 nm)/DNDAPM∶LiQ (50 nm,1∶1)/Yb(1 nm)/Ag(100 nm)。图7 显 示 了EOD 的J-V曲线图,可以看出在2 V 左右电子注入到器件中,DBTRz 和DCNTRz 的电子电流密度大于DTRz。3 种材料都有较好的电子传输能力,且引入缺电子基团二苯基三嗪和氰基,也将改善了主体材料的电子传输和注入性能,其中DCNTRz 的电子传输性能最强。
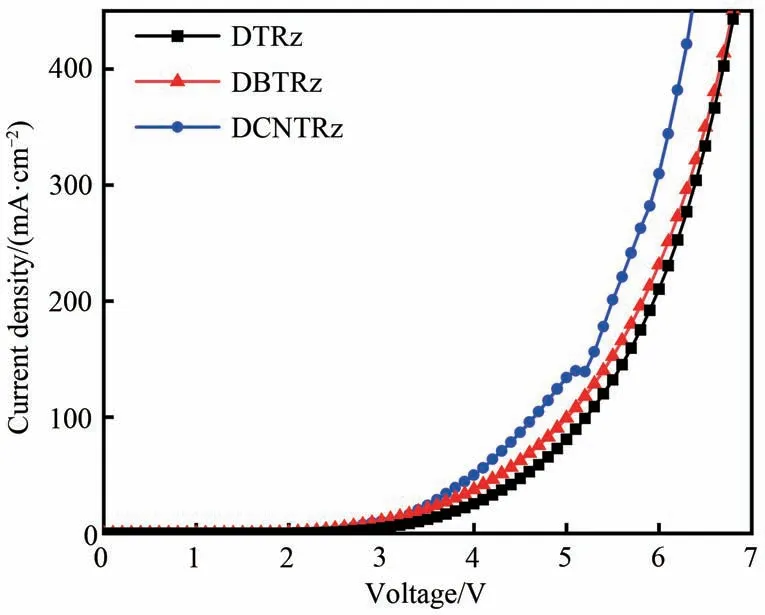
图7 DTRz、DBTRz 和DCNTRz 单 电 子 器 件 的 电 压-电流密度。Fig. 7 Current density-voltage of the electron-only devices for the compounds DTRz,DBTRz and DCNTRz.
3.6 器件结构
三个N 型主体都具有高的热稳定性,适中的电子传输能力,优异的光物理性质等优点,为了方便比较,且氰基和二苯基三嗪均加深了LUMO能级,我们设计了器件结构为:ITO/HATCN(10 nm)/BPBPA(50 nm)/NBPBC∶N-type Host∶Pt(BBP)(40 nm;1∶1∶6%)/DNDAPM∶LiQ(50 nm,1∶1)/Yb(1 nm)/Ag(100 nm)的器件。HATCN(二吡嗪[2,3-f:2',3'-h]喹啉-2,3,6,7,10,11-六碳腈)作为空穴注入层(HIL),BPBPA(N,N,N',N'-四 苯 基 联 苯 胺)为 空 穴 传 输 层(HTL),NBPBC(9,9'-二([1,1'-联苯]-4-基)-3,3'-联-9H-咔唑)为p 型主体,器件I、II 和III 的n 型主体分别为DTRz(器件I)、DBTRz(器件II)和DCNTRz(器 件III),掺 杂 客 体 为Pt(BPP),DNDAPM(2-[4-(9,10-二萘-2-蒽-2-基)苯基]-1-苯基-1H-苯并咪唑)为电子传输层。所有用于器件制备的化合物结构和能级结构如图8 和图9所示。

图8 器件中所用材料的化学结构Fig. 8 Chemical structure of materials used in the device

图9 绿光器件中的能级结构图Fig.9 Energy diagram used in the green PhOLEDs devices
3.7 器件对比
三个器件的电致发光性质如图10 和表2 所示,器件I、II 和III 都具有较低的启亮电压(2.2 V左右)。随着N 型材料LUMO 能级的降低,电子更加容易从DNDAPM 注入到N 型主体材料中,使得器件III 的启动电压比器件I 的降低了0.2 V。在相同电压下,器件II 和器件III 有更高的电流密度,这说明DBTRz 和DCNTRz 中额外吸电子基的引入,增强了发光层的电子传输能力,取得了更好的载流子平衡,从而获得了更高的电流密度。

图10 器件I、II 和III 的亮度-电压-电流密度曲线。Fig.10 Luminance-voltage-current density curves of device I,II,III.
图11 中,器 件I、II 和III 都 展 现 了 良 好 的 器件效率,器件I、II、III 最大电流效率分别是45.8,27.8,46.5 cd·A-1,最 大 功 率 效 率 分 别 是51.4,29.9,45.7 lm·W-1。器件II 的电流效率和功率效率的较低的原因可能是由于多出来的一个二苯基三嗪的空间位阻效应更大,阻碍了主体向客体的能量传递效率。出人意料的是,即使在高亮度下,器件I,II,III仍能保持较高的电致发光效率,在100 cd·m-2,1 000 cd·m-2和10 000 cd·m-2下,其电流效 率分别为42.5,24.2,39.7 cd·A-1,45.8,26.6,46.5 cd·A-1和42.3,27.8,39.8 cd·A-1。3 个材料都表现出了对效率滚降良好的控制,在10 000 cd·A-1高亮度下效率滚降分别是10%、3%和10%。原因可能是N 型主体与P 型主体1∶1 混合后材料拥有较好的空穴和电子的平衡性能。

图11 器件I、II 和III 的电流效率-亮度-功率效率曲线。Fig.11 Current efficiency-luminance-power efficiency curves of the devices I,II,III.
三个器件电致发光光谱如图12,在4 V 电压下的色坐标分别是(0.30,0.64)、(0.31,0.63)和(0.31,0.64)。从客体材料的PL 光谱中也可以看出器件电致发光光谱均为绿光客体的发射光谱,说明激子在主体上形成后,传递给了绿光客体,实现发光。

图12 器件I、II 和III 在4 V 电压时的电致发光曲线和Pt(BPP)在二氯甲烷中的光致发光光谱。Fig.12 Electroluminescence curves of devices I,II and III at 4 V and photoluminescence spectra of Pt(BPP)in dichloromethane.
在4 000 cd·m-2的初始亮度下,我们测试了百分比亮度下降到95%所需的时间,由图13 可以看出,器件III 的寿命(LT95)最长,达到了282 h,比器件I 的寿命(见表2)提高了约54%,器件II 的寿命达到了255 h,比器件I 提高了约39%。由于DCNTRz 有最深的LUMO 能级(-2.23 eV),使得电子更容易注入到主体材料中,避免了电子直接注入到客体Pt 材料中。而Pt 材料本身为富电子构型,电子传递给Pt 客体易导致材料发生不可逆的物理或化学变化,从而影响寿命。
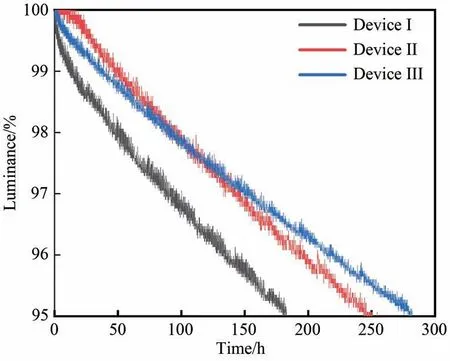
图13 器件I、II 和III 百分比亮度衰减至95%曲线。Fig.13 Life time(LT95)curves of device I,II,III.
4 结 论
通过光物理性质研究表明,我们合成的DTRz、DBTRz 和DCNTRz 绿 光N 型 主 体 材 料 具有较高的三线态能级(2.83,2.71,2.72 eV),与绿色磷光客体材料Pt(BPP)的三线态能级(2.49 eV)相匹配,有效将三线态激子限制在负责发光的客体材料中。通过热力学性能分析3 种化合物均具有良好的热稳定性和形貌稳定性,有利于器件的蒸镀和成膜。用DTRZ、DBTRz 和DCNTRz 制作的器件的启动电压分别为2.3,2.2,2.1 eV,最大电流效率分别为45.8,27.8,46.5 lm·W-1。其中DCNTRz 制作的器件III 最高外量子效率达14.3%,最大亮度达221 589 cd·m-2,在1 000 cd·m-2下电流效率最高达46.5 cd·A-1,功率效率最高达45.7 lm·W-1,通过对比我们发现,对于由P 型和N 型构成的双主体中,N 型主体的LUMO 能级越深,相应的器件寿命越好,其中器件III 在4 000 cd·m-2下器件寿命(LT95)达282 h,比器件I 足足提高了54%。并且,在寿命提高的同时还有14.3%的最大外量子效率和2.1 V 的低驱动电压。综上所述,我们合成的一系列具有较深LUMO 能级的N 型主体材料,实现了绿光器件的低启动电压,长寿命,高效率。
