过渡金属磷酸盐材料NiPS3催化活性密度泛函分析
2022-04-29宋静丽方志刚曾鑫渔朱依文毛智龙刘立娥魏代霞
宋静丽,方志刚,曾鑫渔,朱依文,毛智龙,刘立娥,魏代霞
(辽宁科技大学 化学工程学院,辽宁 鞍山 114051)
随着社会的飞速发展,对能源的开发和使用需求也越来越大。由于二次能源有限,目前处于的紧缺的状态,所以寻求高效环保的新型催化剂成为目前重要科研目标[1]。传统的贵金属催化剂不仅获取困难,且价格昂贵[2]。因此近些年来,价格低廉且易于获取的非晶态合金引起了科研人员的广泛关注[3]。非晶态合金在催化材料[4]、电极材料[5]、磁性材料[6]和光电子材料[7]等方面的运用已经非常广泛。其中过渡金属磷酸盐[8](MTPs)是一种新型二维材料,独特的能带分布和电子结构,使得其具有优异的电子、光学、催化和磁性等物理性质,成为研究热点。
二维层状晶体NiPS3[9]作为众多MPTs材料的代表,具有较多优良性能,被科研人员大量研制并投入实际生活中应用,同时其还是MPTs材料在诱导催化析氢反应中最有潜力的催化剂之一[10]。ZHANG等[11]通过实验发现,NiPS3材料具有显著的电化学稳定性和耐久性,在催化析氢反应中可与商用Pt/C催化剂相媲美,甚至作用优于商用Pt/C。DANGOL等[12]通过液相剥离获得了高产率的超薄2D NiPS3纳米片,并对其多功能电化学性能进行了研究,发现NiPS3是一种优良的水氧化电催化剂。XUE等[13]研究发现,NiPS3-G复合材料可以作为高效电催化产氧(OER)催化剂,因其不含表面活性剂污染且质量负载更低,显示出优于许多传统材料和已报道的很多2D材料的催化性能。LUXA等[14]对NiPS3合成材料在碱性电解质中的析氢反应进行了测试,发现当施加电压脉冲时,其显示出优秀的催化析氢(HER)性能,说明NiPS3材料可成为优良的析氢催化剂。MA等[15]通过第一性原理计算,分析了锂离子在NiPS3材料单分子层上的吸附与扩散,发现锂离子可在Ni和S上强烈吸附,此发现不仅为开发高性能锂离子电池提供了新的机理,也证明了NiPS3可做为候选阳极材料。ZHANG等[16]通过对NiPS3材料的理论计算,发现P和S协同作用下的氢吸附过程的最佳自由能为HER过程提供了巨大的贡献,也为NiPS3材料作二阴离子纳米杂交电催化剂的设计策略制备电催化剂开辟了新的途径。ALAM等[17]基于密度泛函理论,确定了过渡金属三硫化合物NiPS3的单层析氢反应的催化活性位点为一些欠配位的P和S原子,可以为制作贵金属无pH通用电催化剂铺平新道路。
密度泛函理论是一种研究多电子体系电子结构的方法,目前已经广泛应用于分子和凝聚物理及化学性质的研究,例如物质结构及性质、磁性产生、化学催化反应机理、过渡态结构和活化势垒等问题,且对于含有一个或多个过渡金属原子或离子的体系显现出优越性。而团簇则是通过一定的作用力组成的相对稳定的微观聚集体,其可以采用局域化的原子集团模型用以模拟整个晶体在微观上的物质构造,该模型方法是目前研究非晶态材料局部性质的有效方法。
然而,目前对过渡金属磷酸盐材料NiPS3催化性质的研究多停留在宏观层面,缺乏对其微观层面催化性能的产生和作用的具体研究。本文以团簇NiPS3为局域模型,模拟过渡金属里酸盐材料NiPS3微观结构组成,从团簇NiPS3各原子HOMO和LUMO轨道贡献率、能隙差、HOMO和LUMO轨道图、态密度图及库普曼斯定理等方面对NiPS3优化构型的催化作用机理和催化活性进行分析与研究,以期为科研人员设计制备催化活性更高的过渡金属磷酸盐材料NiPS3提供一定的理论参考和支撑。
1 计算方法与稳定构型
1.1 团簇NiPS3的理论模型及计算方法
基于拓扑学原理[18-20]对团簇NiPS3进行研究和分析,以平面五边形、四棱锥型和三角双锥型等为基础构型,运用密度泛函理论[21-23],设计出团簇NiPS3二、四重态下共20种理论初始构型。利用Gaussian09量子化学软件对这20种初始构型进行二、四重态下的全参数优化计算。由于在量子化学计算领域,B3LYP杂化泛函的应用最广泛、综合性能最强,可以对含有过渡金属元素的体系及开壳层结构进行较为准确的计算,def2-tzvp基组可对前四周期元素进行全电子运算,且其计算精度相对较高。因此,构型的优化分析均基于B3LYP/def2-tzvp进行,其中对团簇NiPS3中的P和S加极化函数。团簇NiPS3的HOMO轨道能级(EHOMO)和LUMO轨道能级(ELUMO)、费米能级(EFermi)、能隙差、各构型总态密度图和前线轨道图均利用Multiwfn辅助程序计算得出。所有的数据处理和运行过程均在HP-Z440计算机上完成。
1.2 团簇NiPS3的稳定构型及相对能量
最初设计的团簇NiPS3有20种初始构型,而经过Gaussian09的全参数优化计算和构型分析,排除了相同构型和因存在虚频而不能稳定存在的构型后,最终得到二、四重态下共10种稳定的优化构型,其中二重态下的构型5种,四重态下的构型5种。比较各个构型的校正能发现,构型1(2)的校正能最小,所以构型1(2)相较其它构型更为稳定,因此将构型1(2)所具有的能量作为参考,并将其设为0.000 kJ/mol,其它构型的能量以此为基准重新计算得到相对能量。将各个构型按照其相对能量由低到高的原则,对团簇NiPS3的10种优化构型重新进行排列,如图1所示。
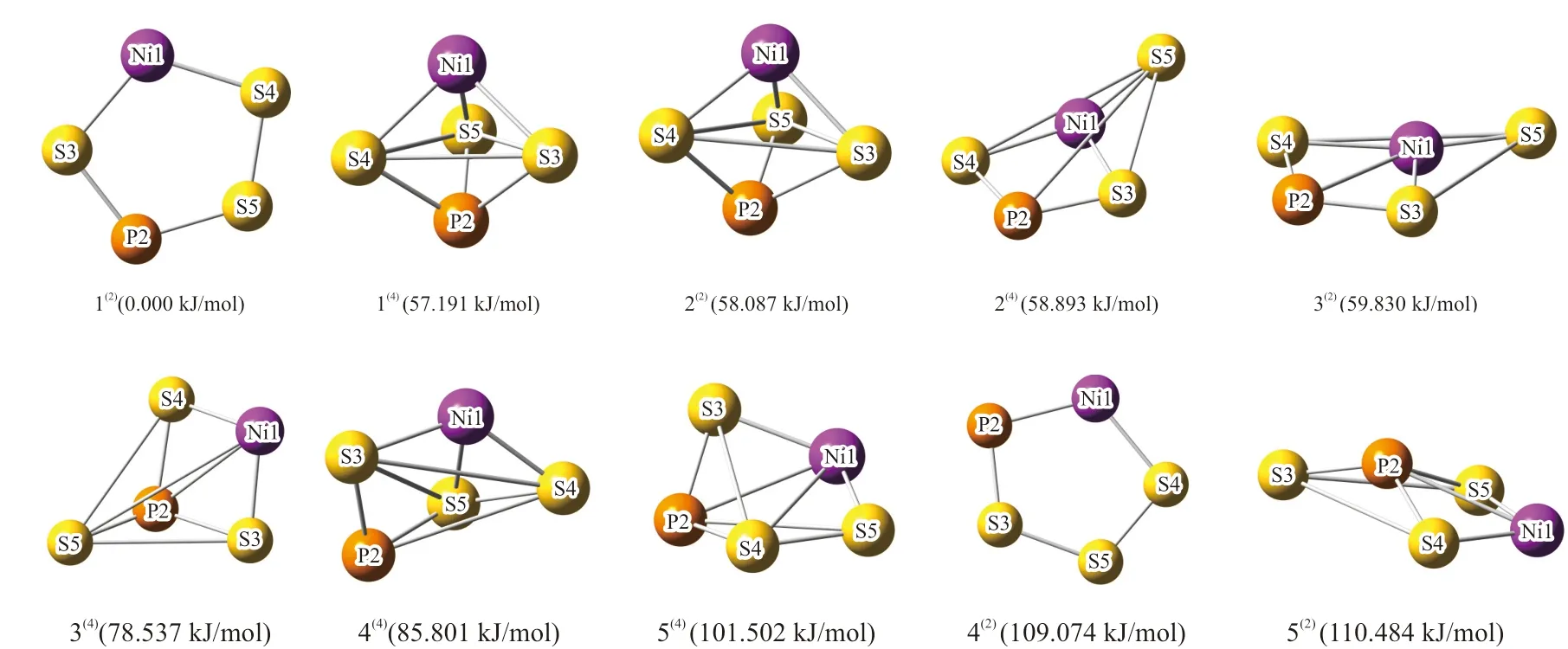
图1 团簇NiPS3的优化构型Fig. 1 Schematic illustration of optimized configurations of cluster NiPS3
由图1可知,团簇NiPS3的初始构型经过一系列构型排除后所得到的优化构型以五边形、三角双锥型、四棱锥型和戴帽三角锥型为主要存在类型。其中平面五边形构型有二重态下的构型1(2)和4(2);三角双锥型构型有二重态下的构型2(2)和四重态下的构型1(4);四棱锥型构型有二重态下的构型3(2)、5(2)和四重态下的构型2(4);戴帽三角锥型构型有四重态下的构型3(4)、4(4)和5(4)。比较各个构型的相对能量大小,可得10种构型按照能量大小顺序为1(2)< 1(4)< 2(2)< 2(4)< 3(2)< 3(4)< 4(4)< 5(4)< 4(2)< 5(2),说明构型1(2)能量最低,相较其它构型更为稳定,构型5(2)能量最高,稳定性最差。
2 结果与讨论
2.1 团簇NiPS3的HOMO和LUMO轨道分析
2.1.1 团簇NiPS3各原子HOMO和LUMO轨道贡献率
20世纪50年代,福井谦一[24]总结出前线轨道理论,研究表明,分子附近的电子云根据能量可细分为不同的能带轨道,电子最高占据分子轨道HOMO和电子最低未占据分子轨道LUMO影响化合物的很多性质,是决定化学反应的关键。前线轨道理论能够通过得出HOMO和LUMO轨道能量、重叠程度和波函数相位等信息,解释一些经典理论无法解释的行为,广泛地应用于化学反应、生物大分子反应过程和催化机理等理论研究。因此,为了更加清晰的探究NiPS3的催化活性规律,找出团簇NiPS3的潜在活性位点,计算得出团簇NiPS3中各原子对HOMO和LUMO轨道的贡献率如表1所示。
由表1可知,团簇NiPS3各个优化构型中Ni、P和S 3个原子对HOMO和LUMO轨道的贡献率均大于0,说明3个原子对团簇NiPS3的催化活性均有贡献。在HOMO轨道贡献率中,除构型4(4)和5(4)是P原子的贡献率最大外,其它构型均是S原子的贡献率最大;在LUMO轨道贡献率中,除构型3(4)和4(4)是Ni原子贡献率最大外,其它构型均为S原子的贡献率最大。由此可推断,在团簇NiPS3中,S原子极有可能是主要的潜在催化活性位点,而此结论也与ALAM等[17]基于密度泛函理论对NiPS3参与HER反应进行分析,所得出的S原子充当活性位点的研究结果相吻合,由此可以预测S原子是析氢反应的潜在催化活性位点。对于团簇不同的重态而言,二重态下的构型均为S原子的贡献率最大,所以二重态下可能只有S原子一个催化潜在活性位点;四重态下,多数构型的前线轨道贡献率仍是S原子最高,在构型3(4)、4(4)和5(4)中,S原子贡献率虽然不是最大,但在HOMO和LUMO轨道总贡献占比也达45%,也验证了S原子极有可能是潜在的催化活性位点这一结论。就单个原子对HOMO和LUMO轨道贡献率的平均值而言,S原子的HOMO和LUMO轨道的贡献率均大于50%,说明在团簇NiPS3分子中,S原子对催化活性的贡献率最大,且非金属原子对催化活性的影响大于金属原子。

表1 团簇NiPS3各原子对HOMO和LUMO轨道贡献率Table 1 Contribution rate of each atom of cluster NiPS3 to HOMO and LUMO orbits
2.1.2 HOMO和LUMO轨道能隙差
HOMO和LUMO轨道能隙差可以表示电子的活泼性和其分布的易变形程度。HOMO轨道能级越高,说明团簇越容易失去电子;LUMO轨道能级越低,说明团簇越容易得到电子。由此可得,HOMO和LUMO轨道能隙差(EGap=EHOMO-ELUMO)的大小体现了电子从HOMO向LUMO跃迁能力的大小,一定程度上也反映了团簇分子参与化学反应催化能力的强弱,即EGap越大,电子从占据轨道向空轨道的跃迁越难发生,团簇NiPS3的催化活性越弱;EGap越小,电子从占据轨道向空轨道的跃迁越容易发生,团簇NiPS3的催化活性越强。因此,团簇NiPS3的催化活性和能力与EGap密切相关。团簇NiPS3的前线轨道能级EHOMO、ELUMO和EGap的数值如表2所示。
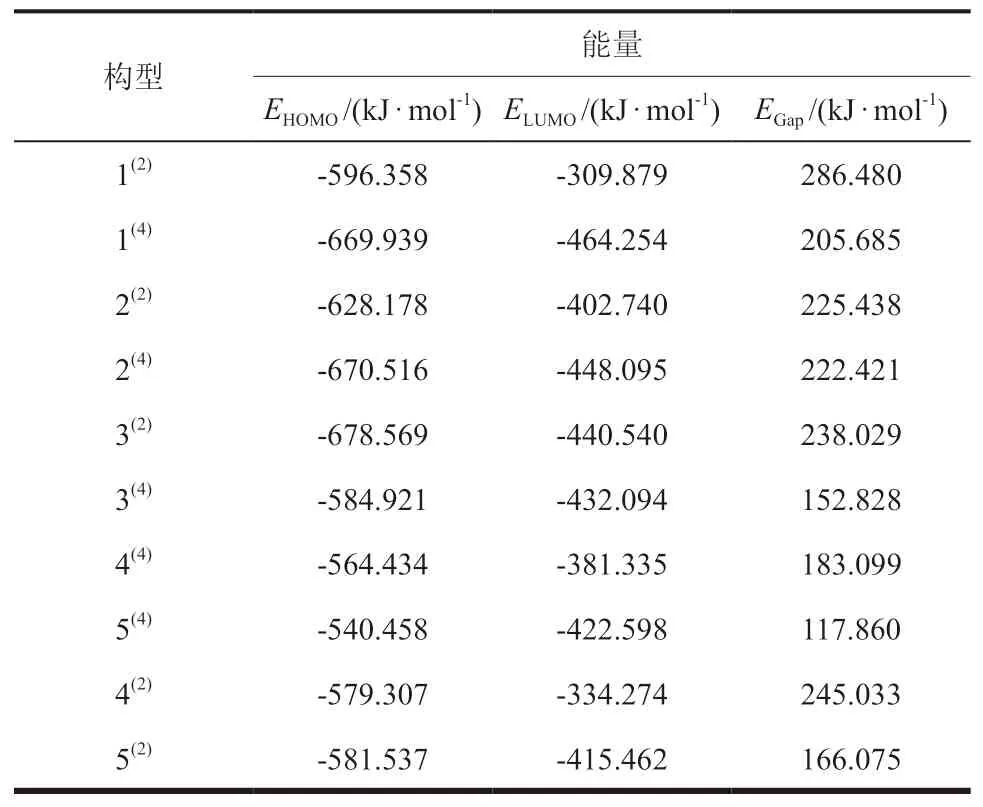
表2 团簇NiPS3的HOMO和LUMO轨道能级和能隙差Table 2 HOMO and LUMO orbital energy levels and energy gap difference of cluster NiPS3
由表2中EGap可知,各个构型EGap大小顺序为:1(2)> 4(2)> 3(2)> 2(2)> 2(4)> 1(4)> 4(4)> 5(2)> 3(4)> 5(4),构型5(4)的EGap最小,构型1(2)的EGap最大,说明在团簇NiPS3中,构型5(4)中电子从最高占据轨道向空轨道的跃迁最容易发生,构型1(2)最难发生,即构型5(4)的催化活性最强,构型1(2)的催化活性最弱。此外,除构型5(2)外,团簇NiPS3二重态构型的EGap均大于四重态构型的EGap,说明相较于二重态而言,四重态优化构型的催化活性更强。
2.1.3 HOMO和LUMO轨道图
由于团簇在化学反应中,HOMO轨道表现出较强的给电子能力,LUMO轨道表现出较强的得电子能力,所以可以通过分析HOMO和LUMO轨道图,了解团簇的得失电子情况,一定程度上能够反映团簇各个构型的化学反应催化活性。团簇NiPS3各个构型的HOMO和LUMO轨道图如图2所示。HOMO轨道图面积的大小表示优化构型的给电子能力的强弱,LUMO轨道图面积的大小表示优化构型得电子能力的强弱。HOMO和LUMO轨道图中各原子附近的不规则阴影区域表示电子在HOMO和LUMO轨道出现时其波函数形成的离域空间,离域空间中电子的出现概率较大,电子流动也相对更加剧烈,红色阴影区域表示相位为负的轨道波函数,绿色阴影表示相位为正的轨道波函数,离域空间与团簇催化活性大小密切相关。
由图2可知,电子在HOMO和LUMO轨道均产生了离域空间,且HOMO轨道图中不规则面积的大小略大于LUMO轨道图中不规则面积的大小,也进一步说明团簇NiPS3具有一定的得失电子能力,且失电子能力略大于得电子能力。进一步分析图2中单个原子附近的离域空间,在团簇NiPS3的各个优化构型中,不规则面积主要集中分布在S原子附近,相对Ni原子和P原子,S原子附近的不规则面积占据了HOMO、LUMO轨道图的大部分区域,这也印证了在上文对团簇NiPS3各个分子HOMO和LUMO轨道贡献率的分析,S原子的贡献率最大,极有可能是团簇NiPS3的主要潜在催化活性位点。

图2 团簇NiPS3各构型的HOMO、LUMO轨道Fig. 2 HOMO and LUMO orbital diagrams of each configurations of cluster NiPS3
2.2 团簇NiPS3态密度图分析
为了方便描述电子在能带中的分布而引入EFermi,其值的大小由EHOMO与ELUMO加和取平均值而得到。EFermi左侧的轨道中有较多电子存在,所以在化学反应中起提供电子的作用;EFermi右侧轨道表示能级因具有空轨道而可以接受电子。提供电子和接受电子的能力均可以反映物质催化活性的强弱,EFermi附近的电子云密度越大,说明团簇越活泼,所以EFermi在一定程度上成为表征团簇催化活性的又一重要参考[25]。态密度(DOS)表示单位能量范围内(E~ (E+ ΔE))的电子状态数目,因此态密度图可以显示出EFerm附近的电子云密度分布情况,一定程度上可以反映团簇NiPS3不同构型的催化活性,EFermi附近的电子云密度大,则该物质的催化活性就越强。由于HOMO轨道代表电子占据轨道中能量最高的轨道,LUMO轨道表示电子未占据轨道中能量最低的轨道,因此可近似认为在EFermi+ ΔE的范围表示LUMO轨道中电子云密度的变化;在EFermi- ΔE的范围表示HOMO轨道中电子云密度的变化。因此,以EFermi为基准,EFermi± ΔE范围内左右波峰的相应情况,可以反映物质的得失电子能力,从而具体显示物质催化活性的强弱。团簇NiPS3各个构型含EFerm的总态密度图如图3所示。由图3可知,在EFermi± ΔE范围内,EFermi左右两侧均存在或大或小的波峰,说明团簇NiPS3在参与化学反应时,既可以给出电子又能够得到电子,具有较强的催化活性,且在10个优化构型中,不论是EFermi± ΔE范围内左右波峰的峰值大小还是峰值高度,EFermi左侧的波峰均大于右侧波峰,由此可知团簇NiPS3具有一定的得失电子能力,但失电子能力略大于得电子能力,这与在上文分析HOMO和LUMO轨道图得出的结论相吻合。
2.3 团簇NiPS3库普曼斯定理分析
基于库普曼斯定理并结合团簇NiPS3各个优化构型HOMO和LUMO轨道的能量数值,可以计算出团簇NiPS3各个构型的电离势(EI,kJ/mol)、电子亲和能(Eea,kJ/mol)、电负性(χ,kJ/mol)和亲电指数(ω,kJ/mol),从而反映团簇不同优化构型催化活性大小的能量参数,相关计算公式如式(1)~(4)所示。

EI表示团簇分子中的原子成为阳离子时所要吸收的能量,是衡量原子对价电子束缚能力的物理量,EI越小,说明越容易失去单子,催化活性越强。Eea表示团簇分子中原子得到电子成为阴离子时所要释放的能量,其大小可以反映团簇分子得到电子的难易程度,Eea越大,说明得电子能力越强,催化活性也越强。χ的大小可以衡量原子对电子吸引能力,团簇分子的χ越大,其催化活性越强。ω表示发生化学反应时团簇分子对电子的接受能力,ω越高,团簇分子的催化活性就越强。计算得出团簇NiPS3各个构型的反应活性参数如表3所示。

表3 团簇NiPS3各构型反应活性参数Table 3 Reactivity parameters of each configurations of cluster NiPS3
由表3可知,EI、Eea和χ的波动范围较为平缓,极大值与极小值的差值较小(EI的极值差为138.111 kJ/mol、Eea的 极 值 差 为154.375 kJ/mol、χ的极值差为113.978 kJ/mol),相较前三者而言,ω的波动更为剧烈,极大值与极小值的差距为1250.632 kJ/mol,波动较大,因此,EI、Eea和χ对团簇催化活性的影响相对较小,ω对团簇催化活性的影响相对较大。具体分析各个构型的反应活性参数值可得,构型5(4)的EI最低,Eea和χ相对较高,ω最高,说明构型5(4)的催化活性最强。这与上文2.1.2节中分析EGap所得出的结论相吻合。
3 结论
(1)本文以NiPS3作为局域模型,利用密度泛函理论计算得出10种优化构型,对所有优化构型分别从各原子对HOMO和LUMO轨道的贡献率、能隙差、态密度图、HOMO和LUMO轨道图以及库普曼斯定理等方面进行了微观层面的分析,发现Ni、P和S 3种原子对前线轨道HOMO和LUMO均有一定贡献,由于S原子对前线轨道的贡献率最大,S原子极大可能是团簇NiPS3的潜在催化活性位点;由于EFermi左右两侧均有波峰出现,左侧波峰的峰值和面积略大于右侧,且HOMO轨道图中的离域面积略大于LUMO轨道图,所以团簇NiPS3具有较强的得失电子能力,且失电子能力略强于得电子能力;构型5(4)的能隙差最小,EI最小,Eea和χ相对较大,ω最大,说明构型5(4)的得失电子优于其它构型,即构型5(4)的催化活性最强。
(2)根据对团簇NiPS3微观催化性能的分析,可以预测S原子极有可能是过渡金属磷酸盐材料NiPS3催化作用的潜在活性位点;构型5(4)的催化性能最好,极大可能是过渡金属磷酸盐材料NiPS3催化反应中的优势构型。
