铀酰配位介导咔唑三羧酸超分子组装体的结构演变
2022-04-28于吉攀胡孔球
王 帅,孟 燎,刘 康,于吉攀,胡孔球,梅 雷
中国科学院 高能物理研究所,北京 100049
晶态多孔材料[1]是一类具有规则孔道或中空笼型结构的固相材料,以无机分子筛[2-3]最为典型。此类材料已经在催化和气体分离等方面具有重要的应用。近年来,基于有机分子骨架的晶态多孔材料也日益受到关注,如金属-有机框架材料(metal-organic frameworks, MOFs)[4-5]、共价有机框架材料(covalent organic frameworks, COFs)[6-8]、氢键-有机框架材料(hydrogen-bonded organic frameworks, HOFs)[9]、多孔有机笼(porous organic cages, POCs)[10-11]和金属-有机配位笼(metal-organic cages, MOCs)[12]等。由于有机结构单元的结构可调性和功能多样性,基于有机分子骨架的晶态多孔材料相比于无机分子筛具有更加丰富多样的结构组成和适应性更广的功能应用。特别地,对于以金属-有机配位键为连接方式的MOFs材料而言,不仅可以对其有机组分单元进行分子结构设计和修饰,还可以对其金属节点进行相应地设计筛选,即根据有机配体配位行为的不同可以选择对应的金属离子与之配位。当采用不同金属中心(如过渡金属、镧系金属或锕系金属离子)时,这些金属离子所呈现的结构形式(单个离子或多离子簇)和理化性质(化学稳定性、催化活性、发光行为等)也会有所差别。有机分子骨架的晶态多孔材料的这些优点,使其在气体分离与存储、催化、传感、生物医药等众多领域具有广泛的应用前景,并吸引了越来越多的研究者对其开展研究。
在众多MOFs材料中,锕系MOFs化合物[13-15]是一类以锕系金属离子作为节点的新兴材料。由于锕系离子所特有的与5f轨道相关的成键特点和化学性质,目前所报道的锕系MOFs材料展现了一些与众不同的结构和性质。例如,在镎(Np)基MOF中,Np离子可以形成由锕酰离子-锕酰离子相互作用(actinyl-actinyl interaction, AAI)连接的无限一维链金属节点或由十八核镎酰构成的镎团簇节点[16];由于高原子序数锕系金属节点对射线的良好阻滞能力,铀或钍基MOFs可以作为一类潜在的功能器件,用于X射线闪烁体[17]和辐射剂量计[18]等领域。然而,与过渡金属基或稀土基MOFs相比,因锕系元素(特别是超铀元素)原料不易获得且一般具有放射性,锕系MOFs材料的研究仍然相对较少。新型锕系MOFs化合物的研究,将为包括种态变化、氧化还原行为和配位化学等在内的众多锕系元素基础研究提供更为详实的化合物结构信息,同时也为拓展锕系功能材料的应用领域、实现锕系核素资源的合理化再利用提供更多参考方案和基础研究数据支持。
本研究团队在前期研究中开展了一系列基于有机芳香多羧酸的锕系MOFs材料研究,包括四苯甲烷四羧酸[19](图1(a))、偶氮苯四羧酸[20](图1(b))、二苯紫精四羧酸[21-22](图1(c))、三苯胺三羧酸[23]等(图1(d)、(e))。利用这些芳香多羧酸作为有机连接子,制备了相应的晶态锕系MOFs材料并对其结构、性质进行相应的表征和分析。例如,四苯甲烷四羧酸铀酰MOF材料具有典型的ctn和bor拓扑结构,这与铀酰离子赤道面上的平面三角形配位环境密切相关;偶氮苯四羧酸铀酰MOF材料具有一维螺旋链的金属节点结构单元,可以通过其分子骨架中的自由二甲铵离子进行离子交换;二苯紫精四羧酸铀酰化合物中的紫精官能团赋予其特殊的光致变色性质,是优良的紫外光探针。
在本工作中,进一步将有机芳香多羧酸体系拓展至咔唑骨架衍生物。咔唑是一种具有稠环刚性骨架结构的含氮芳杂环,分子内含有较大的π电子共轭体系,使得其热稳定性和光稳定性较好。同时,该芳环共轭体系的电子转移能力很强,其衍生物表现出优异的光电性能[24]。拟研究咔唑三羧酸配体H3L(图1(f))与铀酰离子的配位组装过程,利用该配体与铀酰的配位作用调控这一功能配体的超分子组装过程并诱导对应晶态材料的结构演变,合成Carbazole-HOF、Carbazole-UOF1和Carbazole-UOF2三种化合物。对这三种具有不同连接方式的晶态化合物进行单晶结构表征,通过详细的结构对比分析这一配位介导结构演变过程的影响因素,同时,也对其红外光谱进行表征。
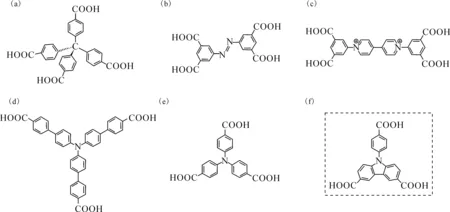
(a)——四苯甲烷四羧酸,(b)——偶氮苯四羧酸,(c)——二苯紫精四羧酸,(d)、(e)——三苯胺三羧酸,(f)——咔唑三羧酸配体(H3L)图1 一些用于锕系MOFs材料合成的有机芳香多羧酸配体Fig.1 Several organic aromatic polycarboxylic acid ligands used in synthesis of actinide-based MOFs
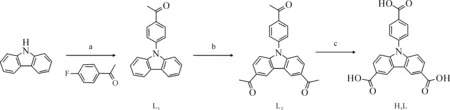
合成条件:a——K2CO3,Cu,DMSO,140 ℃;b——AlCl3,氯乙酰,CH2Cl2,室温;c——液溴,NaOH,p-二氧六环-水,60 ℃图2 咔唑三羧酸配体H3L及其中间体L1与L2的合成示意图[25]Fig.2 Schematic diagram of synthesis procedureof carbazole tricarboxylic acid ligand H3L and its intermediates L1 and L2[25]
1 实验部分
1.1 试剂与仪器
咔唑和4-氟苯乙酮(纯度均为98%),Innochem科技有限公司;Cu粉、二甲基亚砜(DMSO)、乙酰氯、液溴、三氯化铝等其它所用化学试剂均为分析纯,国药集团化学试剂公司。UO2(NO3)2储备液(0.5 mol/L)由六水合硝酸铀酰(UO2(NO3)2·6H2O)溶于超纯水中配制而成。
500 MHz AVANCE Ⅲ型核磁共振氢谱(1H NMR),采用氘代氯仿或DMSO作为溶剂。AmaZon SL Ion-Trap电喷雾质谱仪(ESI-MS)。Tensor 27红外光谱(FTIR)仪,KBr压片,波数范围为400~4 000 cm-1。以上均为Bruker公司。单晶结构测试是在Bruker D8 AVANCE CMOS型X射线衍射仪上采用Mo Kα(λ=0.710 73 Å,1 Å=0.1 nm)或Cu Kα(λ=1.541 78 Å)X射线源完成。
1.2 合成与表征
(1) 咔唑三羧酸配体H3L及其中间体L1与L2的合成
咔唑三羧酸配体(H3L)的合成路线按照文献[25]进行,合成过程示于图2。相关产物的表征结果如下。
配体L1的核磁共振氢谱(500 MHz,CDCl3):δ(ppm)8.21~8.22(d,2H,4-和5-咔唑基H),8.15~8.16(d,2H,2-和6-苯基H),7.71~7.73(d,2H,1-和8-咔唑基H),7.58~7.50(d,2-和5-苯基H),7.42~7.45(m,2H,2-和7-咔唑基H),7.31~7.34(m,2H,3-和6-咔唑基H),2.71(s,3H,苯乙酰基CH3)。
配体L2的核磁共振氢谱(500 MHz,CDCl3):δ(ppm)8.84~8.85(s,2H,4-和5-咔唑基H),8.25~8.27(d,2H,2-和7-咔唑基H),8.12~8.14(d,2H,2-和6-苯基H),7.69~7.70(d,2H,1-和8-咔唑基H),7.45~7.47(d,2H,2-和5-苯基H),2.76(s,6H,咔唑乙酰基CH3),2.73(s,3H,苯乙酰基CH3)。
配体H3L的核磁共振氢谱(500 MHz,d6-DMSO):δ(ppm)12.91(br,3H,COOH),8.98(s,2H,4-和5-咔唑基H),8.25~8.26(d,2H,2-和7-咔唑基H),8.09~8.11(d,2H,2-和6-苯基H),7.83~7.85(d,2H,1-和8-咔唑基H),7.53~7.54(d,2H,2-和5-苯基H)。电喷雾质谱(ESI-MS,Da),m/z(C21H13NO6):理论值,374.07(M-H)-,实验值,373.92。
(2) Carbazole-HOF、Carbazole-UOF1和Carbazole-UOF2的合成
三种基于咔唑三羧酸配体H3L的晶态化合物(Carbazole-HOF、Carbazole-UOF1和Carbazole-UOF2)均通过溶剂热(100 ℃)或水热(150 ℃)反应合成,具体实验条件如下:
Carbazole-HOF:称取19 mg咔唑三羧酸H3L置于15 mL特氟龙反应釜中,依次加入1 mL DMF和1 mL 超纯水溶解配体。继续加入0.20 mL HNO3(4 mol/L)后,将上述溶液混合均匀后密封,100 ℃下反应3 d,然后缓慢降温(3 ℃/h)至室温,得到无色块状晶体。
Carbazole-UOF1:称取19 mg咔唑三羧酸H3L置于15 mL特氟龙反应釜中,依次加入2 mL DMF溶解配体,并与0.10 mL UO2(NO3)2溶液(0.5 mol/L)混合。继续加入0.15 mL HNO3(4 mol/L)后,将上述溶液混合均匀后密封,100 ℃下反应3 d,然后缓慢降温(3 ℃/h)至室温,得到黄色片状晶体。
Carbazole-UOF2:称取19 mg咔唑三羧酸H3L置于15 mL特氟龙反应釜中,依次加入2 mL超纯水溶解配体,并与0.10 mL UO2(NO3)2溶液(0.5 mol/L)混合。继续加入0.20 mL NaOH(1 mol/L)后,将上述溶液混合均匀后密封,150 ℃下反应2 d,然后自然降温至室温,得到黄色棱柱状晶体。
1.3 单晶结构测试与结构解析
使用APEX 3程序收集数据帧,并使用APEX 3程序中的SAINT程序进行数据还原处理。通过SHELXS-97软件的直接方法解析晶体结构,并在Olex2软件中使用SHELXL程序进行全矩阵最小二乘法精修。三种化合物的晶体数据列入表1。本工作中的所有晶体数据已上传并存储于剑桥晶体数据中心(The Cambridge Crystallographic Data Centre, CCDC),三种化合物相应的CCDC编号分别为:2121501(Carbazole-HOF)、2121502(Carbazole-UOF1)和2121503(Carbazole-UOF2)。
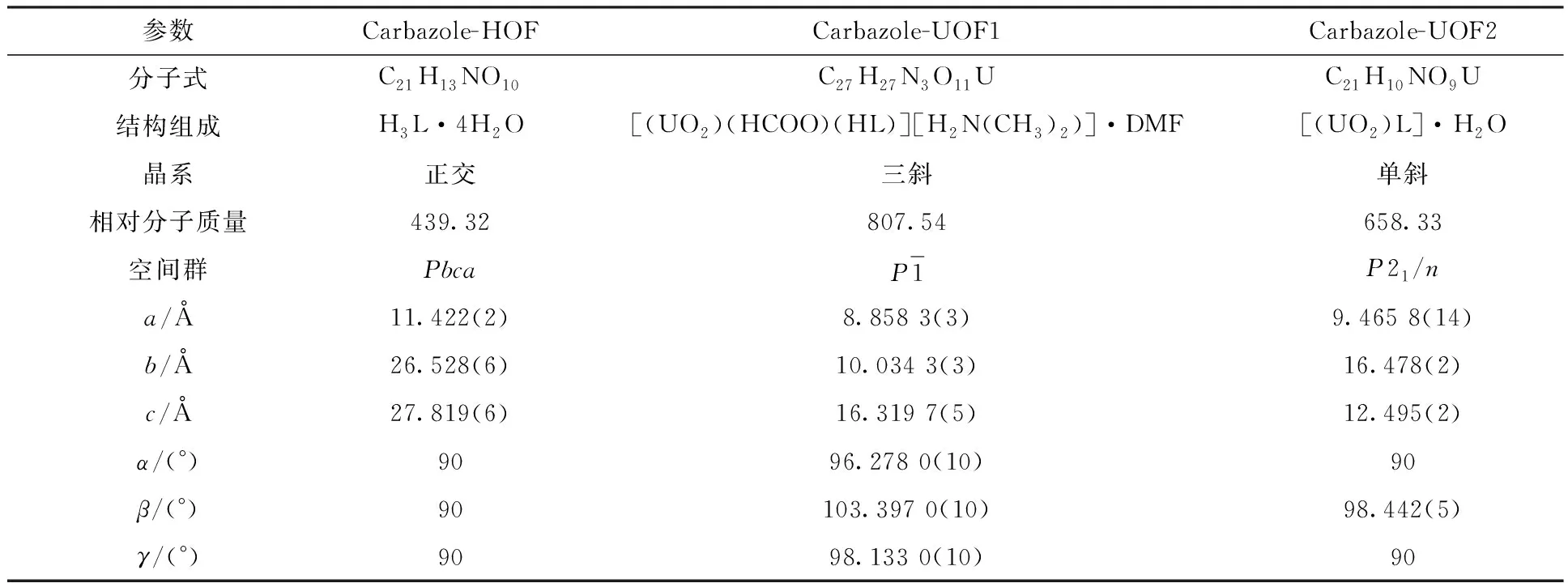
表1 本工作涉及的三种晶体化合物的晶体数据Table 1 Crystal data of three crystalline compounds reported in this work

续表1
2 结果与讨论
2.1 配体及铀酰配合物晶体结构分析
为了与相应的铀酰配合物进行对比,首先研究了在没有金属参与配位时咔唑三羧酸配体自身的晶格堆积方式。咔唑三羧酸配体的结晶产物为Carbazole-HOF,其晶体结构及氢键网络示于图3。由图3可知:所有的三羧酸分子在晶格中均以羧基氢键对的形式与三个相邻分子进行相互作用(图3(a)左图);所有O—H…O氢键的给体O原子和受体O原子之间的距离在2.552~2.750 Å,而H…O的距离为1.787~1.913 Å(表2),具有典型氢键的特征。通过氢键对的二维结构拓展,可以形成类蜂窝型二维氢键有机网络结构,其六方形大孔结构单元的直径约为2.7 nm(图3(b))。值得注意的是,由于配体上的N-苯基与咔唑芳香环平面存在一定的偏转角(52.084°和56.276°,如图3(a)右图),该二维网络结构有别于严格的平面网格,而是呈现波浪形褶皱结构(图3(c)),该二维网络可以进一步通过穿插互锁的“索烃”化方式形成复杂的六重穿插结构(图3(d)、(e))。这一六重穿插结构的形成与其六方单元较大的孔径有关,即大孔径和非平面结构为多重穿插的构建提供了足够的空间,而六重穿插结构中各相邻网络间的堆积作用也有效稳定了大孔径二维网络结构。有意思的是,在发生六重穿插结构后,该晶格中沿a轴方向的孔道结构几乎没有发生变化,依然具有大量的开放孔道结构(图4(a)、(b))。在三维晶格中,这些具有孔道结构的六重穿插单元可以沿着b轴进一步密堆积形成最后的三维超分子框架(图4(c)、(d))。
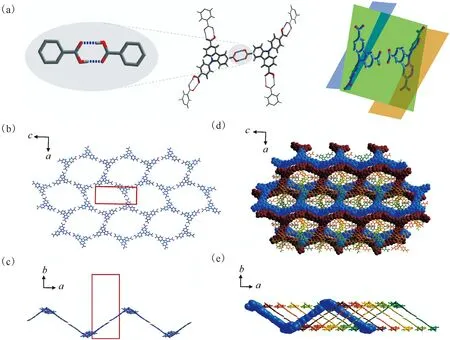
(a)——具有氢键交联网络(左)的咔唑三羧酸结构单元(中)及其非平面分子空间构型(右),(b)——氢键连接的类蜂窝型二维氢键有机网络结构(红色线框表示晶格单胞大小),(c)——二维网络结构的波浪形褶皱结构(沿(001)晶面取向),(d)——二维网络结构间的六重穿插,(e)——六重穿插结构的侧视图(沿(001)晶面取向)图3 Carbazole-HOF的晶体结构及氢键网络Fig.3 Crystal structure and hydrogen bond network of Carbazole-HOF
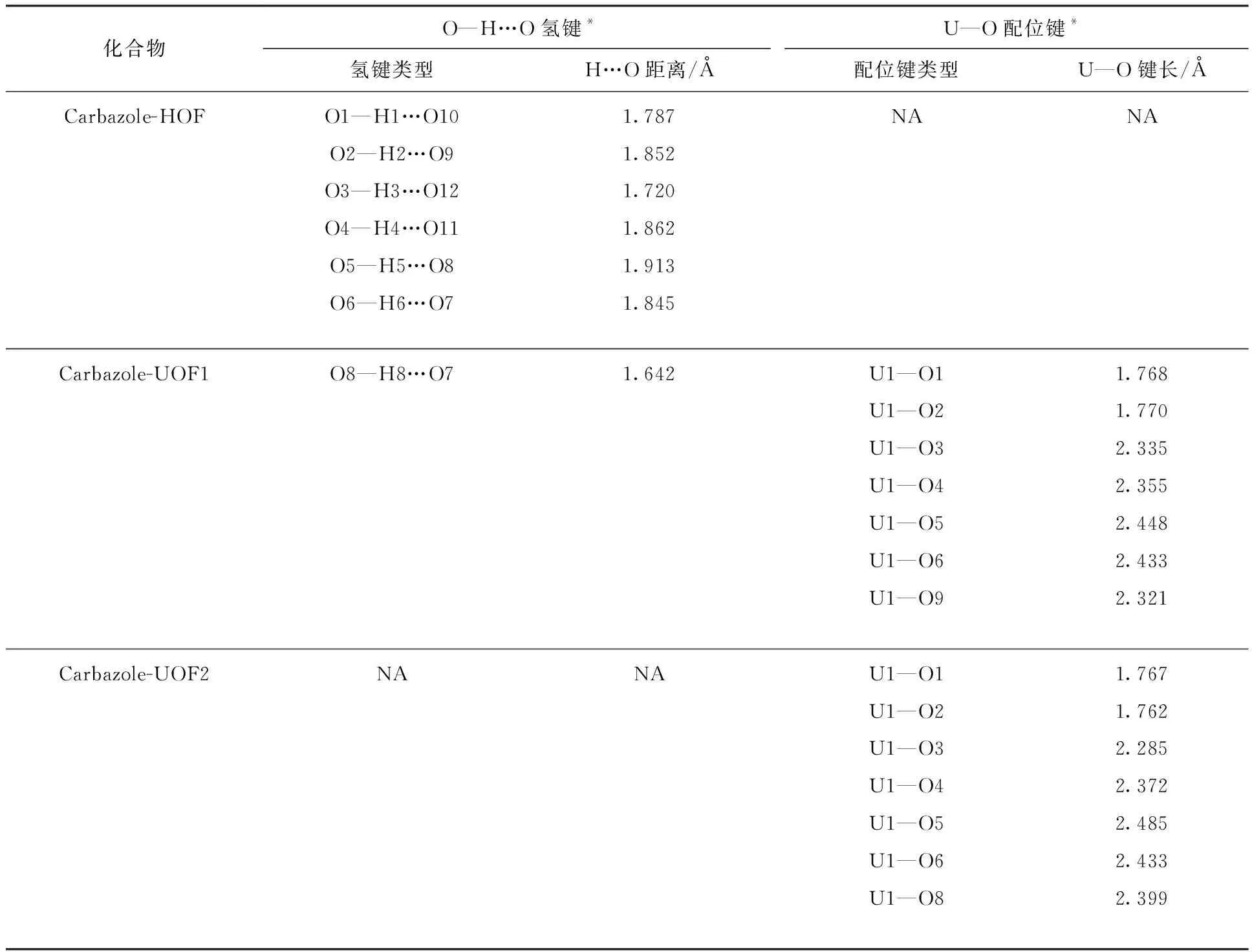
表2 三种化合物中连接不同有机结构的氢键和配位键的参数Table 2 Parameters for hydrogen bonds and coordination bonds that connect different organic structs in three compounds
进一步研究了引入铀酰离子后咔唑三羧酸晶格排列的变化。配位组装反应选择以N,N-二甲基甲酰胺(DMF)为溶剂,利用DMF原位热分解缓慢释放的二甲胺作为碱源来实现对咔唑三羧酸配体中羧酸根基团的可控去质子化。通过咔唑三羧酸和铀酰离子在DMF中的配位组装,成功制备了Carbazole-UOF1。Carbazole-UOF1的晶体结构示于图5。由图5可知:咔唑三羧酸配体的两个羧基分别采取双齿桥联配位和螯合配位的方式与铀酰离子结合,而第三个羧基仍以羧酸形式存在,并与邻近的羧酸基团形成氢键对(图5(a)),这三个羧基分别通过相应的连接形式参与咔唑三羧酸分子之间的组装和晶格堆积。与Carbazole-HOF相比,由于铀酰配位作用替代了部分羧基氢键对,Carbazole-UOF1中有机配体的连接方式发生了较大变化。按照羧基的三种连接方式,可以认为其多元组装过程包括如下几步:首先,一对咔唑三羧酸利用双齿桥联配位的羧基同时与两个铀酰中心配位,形成[(UO2)2L2]双核铀酰结构单元(图5(b));然后这些双核铀酰单元相互之间通过羧基与铀酰螯合配位的方式进一步组装形成一维铀酰-有机链结构(图5(c));最后,通过配位组装形成的一维链进一步通过未配位的自由羧酸基团与其两侧的相邻一维链形成大量氢键对,并最终形成二维超分子网络(图5(d))。值得关注的是,DMF除了作为合成溶剂外,在Carbazole-UOF1的形成过程中也发挥了重要作用。DMF原位分解产生的甲酸根参与铀酰金属中心的配位,使得每个铀酰中心实现了饱和配位,并最终形成五角双锥配位构型(图5(d))。DMF的另一个分解产物二甲胺参与了咔唑三羧酸的羧酸根去质子化,所形成的二甲铵根也以抗衡离子的形式与DMF溶剂分子一起填充在二维超分子网络的孔隙内(图6(a))。另外,这些填充的二甲铵根和DMF分子在二维超分子网络的三维堆积过程(图6(b)—(e))中也通过氢键作用促进了相邻二维网络的交联与堆积。
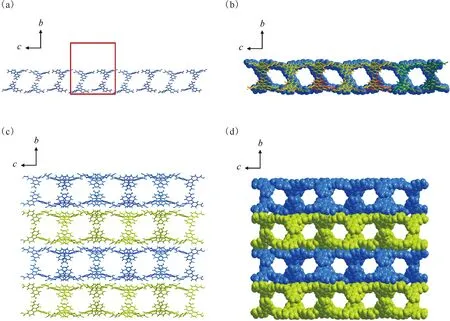
(a)——单个二维氢键网络结构沿(100)晶面取向的侧视图(红色线框表示晶格单胞大小),(b)——六重穿插结构沿(100)晶面取向的侧视图(空间填充模型与棍式模型叠加),(c)——由六重穿插单元沿b轴密堆积所形成的三维超分子框架(分子结构采用棍式模型),(d)——由六重穿插单元沿b轴密堆积所形成的三维超分子框架(分子结构采用空间填充模型)图4 Carbazole-HOF晶格中的开放孔道结构Fig.4 Open pore structure in Carbazole-HOF lattice
当采用NaOH作为碱源时,通过咔唑三羧酸和铀酰离子在水溶液中的配位组装可以得到化合物Carbazole-UOF2。Carbazole-UOF2的晶体结构示于图7。晶体结构分析表明,与Carbazole-UOF1中咔唑三羧酸残留一个未参与铀酰离子配位的羧酸根不同,Carbazole-UOF2的三个羧基均与铀酰进行配位并表现出三种不同的配位模式(图7(a))。更进一步的结构对比表明,Carbazole-UOF2中铀酰结构单元与Carbazole-UOF1铀酰单元仅存在微小差别,即Carbazole-UOF1中的甲酸配位基团(图5)在Carbazole-UOF2中已被咔唑三羧酸的第三个参与铀酰单齿配位的羧基所替换(图7)。这种差别也正与Carbazole-UOF2合成过程中溶液碱性的提高有关,可以有效地实现所有羧酸基团的去质子化并促进羧基后续的铀酰配位过程。从组装过程来看,Carbazole-UOF2的分步组装也仅在最后一步与Carbazole-UOF1有所差别,表现为有别于氢键组装的金属配位驱动组装(图7(b)—(d)),最终形成更为致密的二维铀酰-有机网络结构。从三维堆积方式来看,Carbazole-UOF2的二维铀酰-有机网络与Carbazole-UOF1的二维超分子网络较为相似,均以填充分子(Carbazole-UOF2的填充分子为水分子)参与的层间氢键交联(图8(a)—(d))和π-π相互作用(图8(e)、(f))为主。
2.2 铀酰配位介导咔唑三羧酸超分子组装体的结构演变

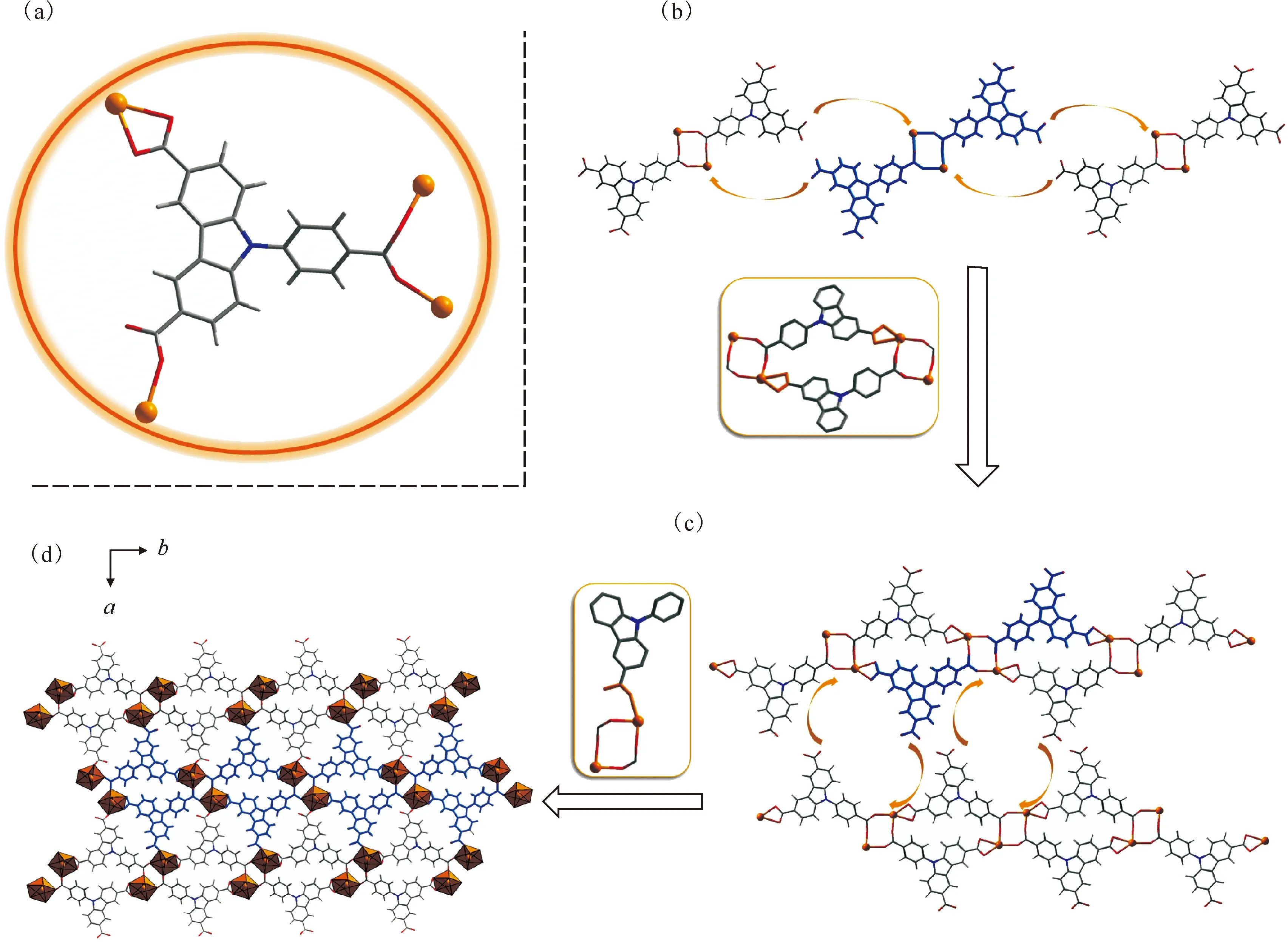
(a)——咔唑三羧酸配体中三个羧基的不同配位模式,(b—d)——从[(UO2)2L2]双核铀酰结构单元(b)到一维铀酰-有机链结构(c)再到二维铀酰-有机网络(d)的逐级组装过程图7 Carbazole-UOF2的晶体结构Fig.7 Crystal structure of Carbazole-UOF2
铀酰配位改变了羧基的连接方式,也推动了咔唑三羧酸组装结构的逐级演变(图10):在没有引入铀酰离子时,每个咔唑三羧酸的三个羧基均参与氢键的形成,通过羧基间的氢键对与邻近的三个配体进行连接,并形成可进行六重穿插的大孔径二维六方网络,即Carbazole-HOF。在加入铀酰离子与部分去质子化三羧酸配体进行配位作用时,去质子化的两个羧基与双核铀酰单元形成一维配位链,并进一步利用第三个未去质子化羧酸基团通过羧基间的氢键对与相邻的一维配位链组装,最终形成Carbazole-UOF1的具有四方孔道的二维超分子网络。当羧酸基团全部参与到铀酰配位时,通过双核铀酰单元的连接作用,Carbazole-UOF1的二维超分子网络将进一步演变为密堆积的二维铀酰-有机配位网络。
在铀酰配位介导结构转变过程中,需要关注铀酰配位构型在其中的重要作用。由于铀酰离子轴向氧原子的占位效应,有机配体与铀酰的配位作用一般发生在其赤道面上,所以铀酰配合物倾向于形成平面结构。在Carbazole-UOF1和Carbazole-UOF2中,由于铀酰离子平面配位构型的影响,HOF结构的波浪形二维曲面结构被拉平为准直性很好的二维平面。而且,铀酰中心的这一构型不同于其它过渡金属中心的配位构型,如Zn基MOF材料中Zn金属节点通常形成双核“风车”型分子构型[25],而Zr基MOF中Zr金属节点一般采取八面体六核簇的构型[26],从而导致形成的UOF与基于过渡金属的咔唑三羧酸配合物在拓扑结构上存在较大差别。这一差别反映了铀酰离子在多孔有机框架材料构筑及结构调控中的独特性,为基于锕系节点的新型无机-有机杂化材料的设计合成提供了更多可能。
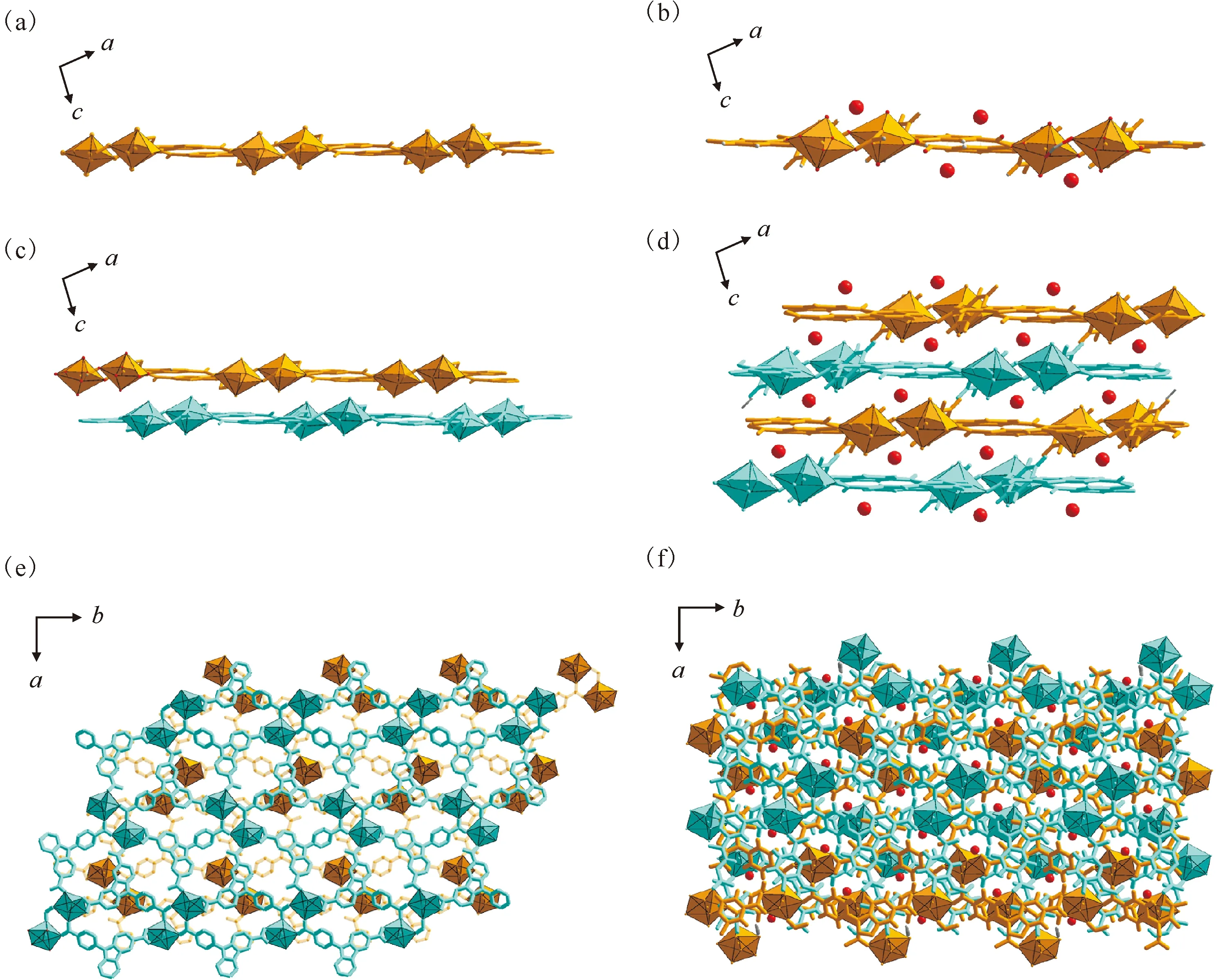
(a、b)——包含填充水分子(a)和不包含填充水分子(b)的单个二维铀酰-有机网络结构沿(010)晶面取向的侧视图,(c、d)——包含填充分子(c)和不包含填充分子(d)的二维铀酰-有机网络在晶格中的堆积(沿(010)晶面取向的侧视图),(e、f)——包含填充分子(e)和不包含填充分子(f)的二维铀酰-有机网络在晶格中的堆积(沿(001)晶面取向的顶视图)图8 Carbazole-UOF2的晶格堆积Fig.8 Lattice packing of Carbazole-UOF2
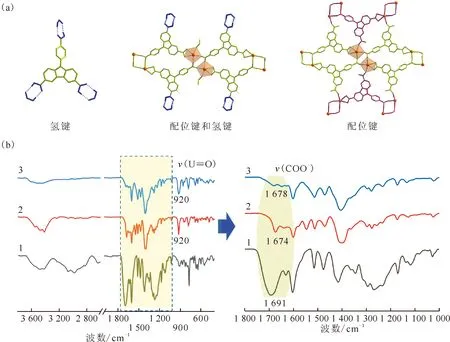
(b):1——Carbazole-HOF,2——Carbazole-UOF1,3——Carbazole-UOF2图9 三种基于咔唑三羧酸配体的晶态材料的结构(a)和红外光谱图(b)Fig.9 Structures(a) and infrared spectra(b) of three carbazole tricarboxylic acid-based crystalline materials
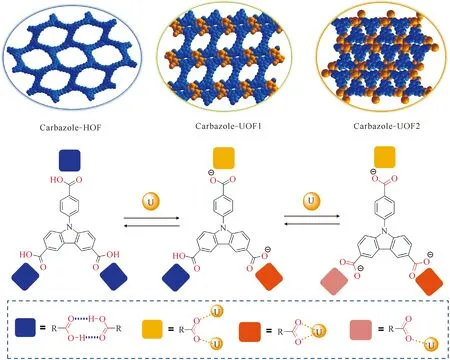
图10 铀酰配位作用介导咔唑三羧酸组装体的结构演变及对应的咔唑三羧酸连接方式Fig.10 Uranyl coordination mediates structure evolution of three carbazole tricarboxylic acid-based crystalline materials
3 结 论
通过引入铀酰金属节点成功地调控了咔唑三羧酸晶格组装方式,实现了从氢键-有机框架结构向金属-有机网络结构的逐级演变。单晶结构表征表明:在这一调控过程中,不同咔唑三羧酸间的连接方式由羧基氢键对逐渐被铀酰配位作用取代,二维网络结构也从六重穿插非平面网络向平面超分子网络和配位网络转变。相关转变过程主要表现在羧基不断参与铀酰中心的配位作用,这一过程也得到了红外光谱的证实。本工作的开展揭示了铀酰-配体配位键合在无机-有机多孔材料制备合成与结构调控中的关键作用,为发展基于锕系金属节点的新型多孔晶态配合物和相关功能材料提供了重要参考。未来将在此类锕系-有机材料的性质表征和功能应用上开展更多相关工作。
