一家系2例Joubert综合征的临床特点和CPLANE1基因变异分析*
2022-04-27薛永珍李培美孙贞超胡艳艳
邹 莉 马 岭 薛永珍 李培美 孙贞超 胡艳艳
1 山东省临沂市人民医院儿科 276003; 2 平邑县中医医院儿科; 3 临沂市人民医院影像科
Joubert综合征(JS,OMIM213300)是一种罕见遗传病。发病率为1∶80 000~1∶100 000[1]。JS临床表现多样,主要临床表现为肌张力低下、呼吸节律异常、眼部运动障碍、共济失调和发育迟缓,亦可合并其他表现。JS的临床异质性与其显著的遗传异质性密切相关,到目前为止,已经发现至少35个基因与JS相关,大多数是常染色体隐性遗传,最常见的致病基因有TMEM67、CPLANE1、CC2D2A等,仅一个少见基因OFD1表现为X连锁隐性遗传[2]。本文中,笔者应用包含JS相关基因的二代测序Panel检测来自同一家系中2例JS患儿,并通过一代测序对家系中其他成员进行验证,以明确其病因,分析探讨CPLANE1基因突变引起JS发病的分子机制及其与表型相关性。
1 病例资料
1.1 先证者 男,3个月,2020年4月出生,因3个月竖头差于2020年7月首次就诊。患儿系G4P4,足月顺产,出生体重3.85kg。生后无缺氧、窒息。生后因“呼吸窘迫”在当地医院住院5d。黄疸2周消退。生后1个月因“呼吸急促”以“重症肺炎”在本院住院7d。至今3个月竖头差,不追视。家族史:父母非近期结婚,身体健康,有3个姐姐,大姐、三姐身体健康,二姐症状与先证者相似。
查体:神志清,精神可。全身皮肤黏膜未见皮疹、黄染及出血点,全身淋巴结未触及肿大。头颅无畸形,毛发分布正常。前囟平坦2cm×2cm。双眼斜视,水平震颤,不能注视,双侧瞳孔等大等圆,对光反射灵敏。口唇无发绀。心肺腹查体未见明显异常。专科情况:寻声追视差。仰卧位肢体对称,拉起头后垂,不能翻身,俯卧位短暂抬头,全前倾坐,立位不负重,四肢肌力正常,肌张力低。双侧膝跳反射存在。血常规、尿常规、肝肾功能、生化、心肌酶、血尿遗传代谢病筛查、脑电图结果均正常。Gesell发育商评估水平为50。双侧眼底检查无异常。颅脑MRI:小脑蚓部体积缩小,双侧小脑上角增宽,呈“磨牙征”,四脑室扩张,形态失常,小脑半球见“中线裂”征。见图1。
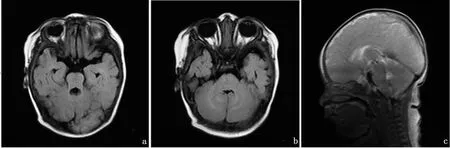
图1 先证者颅脑MRI表现 a. 轴位T2-FLAIR示“磨牙征” b.轴位T2-FLAIR示“中线裂”征 c.矢状位T2WI示小脑上脚增宽,呈前后方向走行
1.2 先证者二姐 女,5个月,2013年9月出生,因5个月竖头差于2014年2月首次就诊。患儿系G2P2,足月顺产,出生体重3.55kg。生后无缺氧、窒息。生后以 “新生儿感染”在当地医院住院5d。黄疸2周消退。生后1个月因“呼吸急促”以“支气管肺炎”在当地医院住院6d,其间查心脏超声提示房间隔缺损。生后5个月竖头差,不会翻身。查体:神志清,精神可。全身皮肤黏膜未见皮疹、黄染及出血点,全身淋巴结未触及肿大。头颅无畸形,毛发分布正常。前囟平坦2cm×2cm。双眼斜视,水平震颤,不能注视,双侧瞳孔等大等圆,对光反射灵敏。口唇无发绀。心肺腹查体未见明显异常。专科情况:追视差,寻声可。不能主动抓握。仰卧位肢体对称,拉起头后垂,不能翻身,俯卧位抬头<45°,全前倾坐,立位不负重,四肢肌力正常,肌张力低。双侧膝跳反射存在。血常规、尿常规、肝肾功能、生化、心肌酶、血尿遗传代谢病筛查、脑电图结果均正常。Gesell发育商评估水平为45。双侧眼底检查示可见视盘边界清。先证者二姐初次就诊时,颅脑MRI报告提示未见明显异常,家长曾辗转多地就诊,均未明确诊断,自行在家未予特殊治疗。先证者就诊时,二姐7岁扶走,仍不能独走,追溯二姐病史,调取既往影像学资料,再次请影像科阅片发现颅脑MRI显示小脑蚓部体积缩小,双侧小脑上角增宽,前后方向走行,呈“磨牙征”,四脑室形态失常,呈“蝙蝠翼状”,二姐的MRI表现较先证者重。见图2。
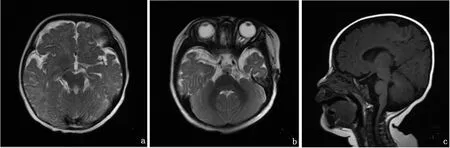
图2 先证者二姐颅脑MRI表现
2 基因检测
为明确患儿分子学诊断,征得患儿父母知情同意下,采集2例患儿及父母、姐妹的外周血各2ml提取基因组DNA。分子遗传学检测结果显示,在先证者和先证者二姐的CPLANE1基因均存在c.1270C>T(NM_023073;exon10)以及c.8901C>A(NM_023073;exon48)两个杂合变异,Sanger测序结果显示,c.1270C>T位点杂合变异来源于父亲,c.8901C>A位点杂合变异来源于母亲,先证者大姐存在c.1270C>T位点杂合变异,先证者三姐存在c.8901C>A杂合变异。见图3。父源性变异在1270号核苷酸由胞嘧啶C变为胸腺嘧啶T,导致氨基酸改变(p.R424X),发生无义突变。母源性变异在8901号核苷酸由胞嘧啶C变为腺嘌呤A,导致氨基酸改变(p.Y2967X),发生无义突变。在dbSNP、1000G、HGMD、Pubmed等文献数据库均未有两个位点的报道。根据美国医学遗传学与基因组学学会(ACMG)指南判断两个变异为致病性变异,致病等级分别为PVS1+PM3_Strong+PM2,PVS1_PM4+PM2+PM3。结合2例患儿的临床表现,因此判断CPLANE1基因复合杂合突变是导致JS的原因,复合杂合突变分别遗传自父母,父母和先证者大姐、三姐均为杂合子,无临床表现,符合常染色体隐性遗传的模式。
3 讨论
JS的诊断需要3个主要标准:(1)影像学的“磨牙征”;(2)婴儿期肌张力低下并伴有迟发性共济失调;(3)发育迟缓/智力障碍。其他常见特征还包括异常的呼吸模式和眼球运动。除上述表现以外,JS常合并其他症状,包括视网膜发育不良、肾脏疾病、视神经缺损、枕叶脑膨出、肝纤维化、多指和或其他异常症状,以区别于单纯型JS无上述其他症状。JS特征性的影像学MRI表现包括“磨牙征”、“中线裂征”、“蝙蝠翼状”及“三角形”第四脑室等,其中“磨牙征”是诊断本病最重要的特征[3]。JS患儿普遍存在运动发育落后,而认知能力有差异。据报道JS患儿全量表智商为(64.3±15.3)[4]。小脑蚓部发育不良程度与认知障碍密切相关[5]。JS患儿婴儿期即出现肌张力低下、眼球震颤、动眼神经失用和异常呼吸模式,共济失调随着时间的推移而进展,眼球震颤随着年龄的增长而改善,异常呼吸模式可表现为交替性呼吸急促、呼吸暂停,有些婴儿死于呼吸暂停,但呼吸暂停随着年龄的增长而改善。在本研究中,该家系姐弟2例JS患儿临床均表现为肌张力低下、发育迟缓、眼部运动障碍、磨牙征和阵发性呼吸急促病史,无其他合并症,符合JS的诊断标准,且为单纯型JS。姐弟2人发育商评分分别为45和50,姐姐小脑蚓部发育不良程度比弟弟重,与上述报道结果一致。
JS的诊断基于特征性临床表现和MRI表现,但症状和体征在个体中各不相同,甚至在同一个家庭的不同成员中也会不同[2]。仅通过临床表型和影像学诊断可能容易漏诊,还需要JS的分子学诊断。已报道的单纯型JS相关基因有CPLANE1、CEP104、ARMC9、ARL13B、KATNIP、B9D1[2]。CPLANE1基因也称为C5ORF42,定位于染色体5p13.2,编码纤毛发生和平面极性效应因子1蛋白。在CPLANE1变异患者中,多为单纯神经系统表型,少数表现为多指,均无肾脏、肝脏、视网膜疾病表现,智力障碍也较其他类型JS患者轻[6]。而且,神经影像学改变较其他类型JS轻微,容易漏诊[7]。MRI检查需要进行多切面扫描来发现不明显的磨牙征和小脑蚓部发育不良,即使是很有经验的放射科医生也可能错过JS的影像学标志[2]。本次报道的JS姐弟为单纯神经系统表型,影像学表现轻微,早期对JS的认识不足,导致既往未发现姐姐的影像学表现,通过此次报道能提高临床医生和影像学医生对JS的认识。
综上所述,JS是罕见的神经发育障碍疾病,具有显著的临床和遗传异质性,本研究中姐弟2人CPLANE1基因存在致病性复合杂合变异,诊断为JS17型,无肝、眼、肾等脏器受累,为单纯型JS,CPLANE1基因变异位点国内外尚未见报道。CPLANE1基因突变型的JS影像学容易漏诊,通过此次报道为临床医生和影像学医生提供更多的诊疗经验,扩展了CPLANE1基因突变谱,同时为JS的病患家庭提供产前诊断具有重要临床意义。
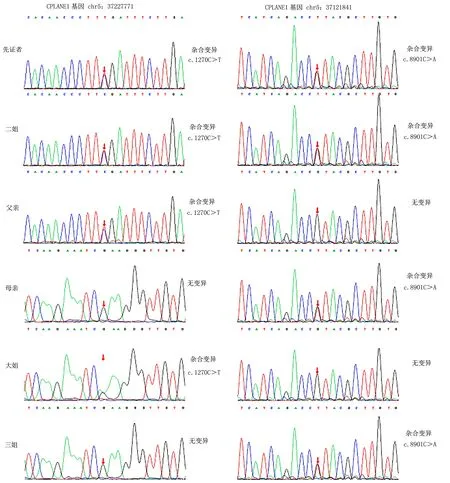
图3 Sanger测序图
