竹原纤维制备过程各阶段形态结构及理化性能分析
2022-04-27林晓英李立轻
林晓英,齐 飞,丁 倩,汪 军,李立轻
(1.东华大学,上海 201620;2.上海海关工业品与原材料检测技术中心,上海 200135)
0 引言
随着绿色环保理念的普及,天然纤维的应用领域备受关注。竹原纤维作为天然可再生纤维素纤维,是功能性良好的纺织新材料,具有广阔的开发应用前景[1-2]。但由于竹原纤维横跨纺织业和林业两大行业,目前行业管理部门以及科技界仍未对竹原纤维及其相关产品给予明确定位。纺织行业竹原纤维相关检测的国家标准尚未建立,仅在2012年、2016年发布有地方性标准。同时,因国内经营和国际贸易方式尚未规范化,市场上存在以麻类纤维替代竹原纤维或以次充好的问题。已有文献研究结果表明,对竹材的检测方法单一,且关于竹原纤维的纵向检测研究不多,多为与其他几种天然纤维素纤维的横向对比[3]。因此笔者采用扫描电镜法、红外光谱分析法、X射线衍射法、热重分析法、X射线光电子能谱(XPS)分别对竹原纤维制备过程中各阶段样品进行纵向的综合检测,研究对比各阶段样品的理化性能和形态结构,以描述在制备各阶段试样的基础性表征。
1 试验
1.1 试验材料
竹原纤维各阶段试样由江苏某公司提供。分别为:① 经过初步处理的第一阶段竹丝,记为阶段1试样,用a表示;② 经过二次工艺处理后的竹丝,记为阶段2试样,用b表示;③ 最终的竹原纤维,属于工艺纤维,记为阶段3试样,用c表示。各阶段试样宏观形态见图1。

a) b) c) 图1 各阶段试样宏观形态
1.2 仪器
傅里叶变换红外光谱仪(Spectrum Two),D/max-2550 PC型18 kW转靶X射线衍射仪(XRD),TG209F1 Libra型热重分析仪(TG),Escalab 250Xi型X射线光电子能谱仪(XPS),DXS-10ACKT型扫描电子显微镜(SEM),光学显微镜。
2 检测结果与讨论
2.1 傅里叶变换红外光谱结果与分析
利用傅里叶变换红外光谱仪(Spectrum Two)测试竹原纤维各阶段的红外光谱图,见图2。

图2 竹原纤维各阶段的红外光谱图
由图2可知,在4000 cm-1~500 cm-1范围内,竹原纤维各阶段试样出现吸收峰的位置一般都在相同的基团振动范围内,且峰形基本相同。各阶段试样都在3400 cm-1~3200 cm-1附近出现O-H键伸缩振动纤维素特征强吸收峰,在2900 cm-1附近出现C-H键伸缩振动中强度吸收峰,在1637 cm-1附近的吸收峰来自试样中吸收水的弯曲振动,在1425 cm-1处小吸收峰来自CH2对称弯曲振动,在1367 cm-1附近的吸收峰来自木质素芳香环中C=O伸缩振动,在1155cm-1处吸收峰来自纤维素的C-O反对称伸缩振动,在1025 cm-1处吸收峰来自纤维素的C-O伸缩振动。但竹原纤维制备过程各阶段试样的差异主要表现在:阶段1试样a与阶段2试样b在1260cm-1处出现木质素苯基醚键的C=O伸缩振动,而阶段3试样c在此附近吸收峰消失,可能是由于木质素中碱溶木质素的降解所引起, 表明与羰基共轭的芳香族核侧链发生了断裂或消失[4]。同时,阶段3试样c在804 cm-1附近吸收峰消失,此处为木质素S,H环上取代基特征峰代表,可能是由于部分碱溶木质素的降解所致,也表明纤维制备工艺过程中木质素发生了部分溶解[5]。
2.2 X射线衍射的结果与分析
采用D/max-2550 PC型18 kW转靶X射线衍射仪(XRD)测试竹原纤维各阶段的X射线衍射对比见图3和表1。

图3 竹原纤维各阶段的X射线衍射对比

表1 竹原纤维各阶段的X射线衍射数据对比
由图3的X射线衍射对比和表1的X射线衍射数据可看出,竹原纤维各阶段试样的峰形和峰位未发生明显变化,衍射曲线的衍射峰分别出现在约16°,22°和35°。说明竹原纤维在制备过程中并未改变天然竹纤维的结晶结构与属性,基本归属于纤维素Ⅰ型晶体[3]。经计算,竹原纤维各阶段的相对结晶度呈依次递增趋势,分别为39.49%,42.37%和46.36%。可能是因为竹原纤维在制备过程中部分胶质物质被溶解,纤维素分子空间增加,一些原来无序排列的微纤丝有序重排,使定形区增加,因而相对结晶度存在增加趋势[4]。
2.3 热重的结果与分析
利用TG209F1 Libra型热重分析仪(TG)测试竹原纤维各阶段的TG特征数据,见表2;竹原纤维各阶段样品的TG对比和DTG对比见图4、图5。

表2 竹原纤维各阶段的TG特征数据
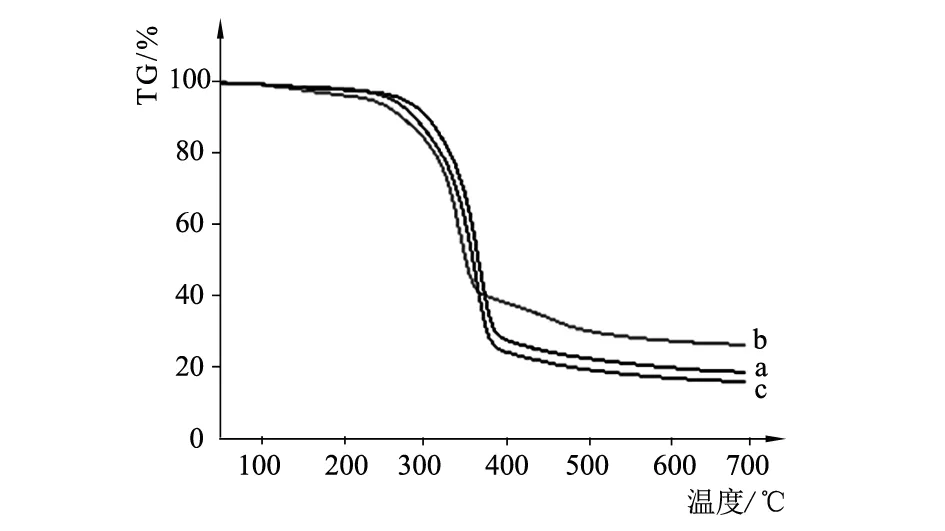
图4 竹原纤维各阶段样品的TG对比

图5 竹原纤维各阶段样品的DTG对比
由表2与图4、图5可知,竹原纤维制备各阶段样品的热失重过程主要分为3个阶段:① 温度在100℃~200℃时,失重十分缓慢,基本无变化,部分损失是由于纤维受热后水分被蒸发引起的;② 温度在200 ℃~400 ℃时,该阶段是竹原纤维各阶段试样热分解主要阶段,均出现最大失重以及失重速率极值峰,阶段2试样b略早于阶段1试样a和阶段3试样c开始分解,其热分解是最早结束的;③ 400 ℃之后,该阶段主要为剩余的木质素热解炭化,失重平缓并逐渐趋于零值。各阶段试样的残余质量分别为18.72%,26.46%,16.00%。残余质量的不同,一方面与各阶段初始含水率差异有关,另一方面由各阶段试样主成分的热稳定性差异所致[6]。
2.4 XPS结果与分析
利用Escalab 250Xi型X射线光电子能谱仪(XPS)测试竹原纤维各阶段试样表面元素含量对比见表3;各阶段试样的C1s相对含量见表4。

表3 各阶段试样表面元素含量对比

表4 各阶段试样的C1s相对含量
结合表3的表面元素含量及O/C比,阶段2试样b表面的Si元素含量增加较大,且 O/C比也增大,可能是因为从阶段1试样a到阶段2试样b过程使用柔软剂所致,从而改变表面元素含量[7]。由表4可知各试样均存在纤维素4种不同的碳键[8],C-C表示纤维表面木质素和抽提物的含量,经工艺处理后,C-C相对其他的碳含量由78.21%变成73.33%,说明在竹原纤维制备过程中有木质素降解及结构变化。化合物则主要体现在C-O和O-C-O/C=O上,阶段3试样c的碳水化合物含量增加,意味着纤维表面暴露出了更多的亲水性基团,这些亲水性基团有利于纤维氢键结合的形成[9]。因此,这个结果较好地解释了竹原纤维物理强度性能显著提高的原因。
2.5 表观形貌特征分析
用光学显微镜和扫描电子显微镜观察竹原纤维各阶段纤维试样的横、纵向形态结构特征,观察结果见图6。其中,a),c),e)为光学显微镜显示;b),d),f)为扫描电子显微镜显示。试样胞壁厚度和细胞中腔直径的测试结果分别见表5和表6。

a) b)

c) d)

e) f) 图6 竹原纤维各阶段试样的横纵向形态结构

表5 试样胞壁厚度的测试结果

表6 试样细胞中腔直径的测试结果
由图6的显示形态可以看出:试样中的单纤维横截面均近似圆形且有裂纹,内部呈天然中空,且中腔多为圆形;纵向有横节和沟槽,表面粗糙且覆盖大量的木质素,有胶质黏连,粗细不均匀。这是因为制备过程中试剂可以松解纤维束间的交合或去除初始微纤维间的木质素,使纤维形态趋于规则,结构精致,表面光滑,甚至出现发光结构[10]。表5和表6结果表明,在制备过程中,竹原纤维的壁厚变化不大,阶段3试样c的中腔有一定程度的扩大,说明该过程纤维出现轻微溶胀。
3 结论
3.1从微观形态看,阶段3试样c为工艺纤维,其制备过程的各试样横、纵向形态特征基本相同,其中竹单纤维均呈圆形截面,有中腔且中腔有微小的溶胀趋势,纵向有胶质黏连。
3.2从物理结构看,竹原纤维制备过程并未改变天然竹纤维的结晶结构与属性,但结晶度呈递增趋势。
3.3从化学结构看,竹原纤维制备过程各试样的主要官能团结构无明显变化,但该过程木质素的降解使1260 cm-1和804 cm-1吸收峰消失。制备过程中试剂的使用,改变了纤维表面的化学变化,C-C 相对其他的碳含量由原78.21%降为73.33%,发生木质素的降解和部分结构变化。
