液态铅铋介质中奥氏体不锈钢腐蚀第一性原理研究
2022-04-20赵永福
熊 静 邓 平 高 军 赵永福
(中国核动力研究设计院,四川 成都 610213)
1 概述
液态铅铋合金(LBE)的高导热性和高热容等优异性质,使得其在加速器驱动次临界系统(ADS)和铅冷快堆领域作为冷却剂具有潜在应用前景[1-2]。奥氏体不锈钢(AS 钢)具有良好的高温力学性能等,已成为反应堆当前的主要候选结构材料。然而,AS 钢在液态铅铋合金介质中的腐蚀现象,将直接影响核反应堆的安全运行,引起了人们的广泛关注。
目前LBE 与金属相互作用研究主要通过环境试验进行,获得的材料腐蚀速率试验数据以及腐蚀后材料微观表征试验结果可以揭示材料的服役性能以及冷却剂化学工况对材料腐蚀特性的影响。然而,虽然相关试验研究在一定程度上揭示了材料在LBE 中的腐蚀机理和规律,但是高温高压试验成本较高,且环境试验缺乏基于原子、分子层面腐蚀机理机制的研究和探索,对材料在LBE 环境下腐蚀的本质认识不够。因此,有必要开展奥氏体不锈钢与液态铅铋介质相容性第一性原理研究,从微观层面探究腐蚀过程中原子的扩散行为和原子间相互作用行为。
本研究拟采用第一性原理计算的方法获得AS 不锈钢、PbBi 超晶胞原子结构模型和表面的能量,以及AS/PbBi 界面的界面能和界面结合强度,从原子及分子尺度揭示AS 钢在铅铋介质中的腐蚀性能和微观腐蚀机理,为铅铋堆的相关实验和设计提供一定的理论指导,因而具有重要的科学意义和应用前景。
2 计算方法和原子结构模型
2.1 计算方法
为开展AS 不锈钢在铅铋介质中的腐蚀特性研究,需要分别构建AS 不锈钢、PbBi 介质的超晶胞原子结构模型,并基于此分别获得其具有较低能量的表面原子结构模型。
在超晶胞原子结构模型构建时,模拟计算分为两个主要步骤:结构优化和静态驰豫。为了保证结果可靠性并降低计算复杂度,这两个步骤要采用不同的电子轨道处理方法和收敛精度。在完整的超晶胞结构模拟时,对于结构优化计算,电子步迭代的能量收敛精度设置为10-5eV,离子步迭代的力收敛精度为10-3eV/Å,并采用Methfessel-Paxton 方法来处理电子对轨道的占据问题;而对于静态电子优化,电子步和离子步迭代的能量收敛精度分别为10-6eV 和10-5eV,并采用具有更高精度的Blöchl- 四面体法来处理电子对轨道的占据问题。
对于表面结构计算,除了结构优化中离子步力收敛精度已经不适用需要更换为能量收敛精度10-4eV,其他设置均与完整晶体模拟保持一致。
2.2 超晶胞原子结构模型
AS 不锈钢的原子结构模型参考316 不锈钢(一种AS 不锈钢)的组分比例构建而成,考虑了材料中含量较高的Fe、Cr 和Ni 三种元素。通过综合考虑,本计算工作采用的AS 不锈钢超晶胞原子总数为32,即铁、铬和钼原子数分别为22、6 和4。运用能量最低的原则,对AS 不锈钢所有可能的原子结构进行了计算,优化后获得a = b= c,α = β = Y = 90°的面心立方结构模型,其原子结构模型如图1 所示。表1 列出了实验和本计算所采用的AS 不锈钢的化学成分,从表中可知,在模拟中的质量比和原子个数比均和实验的质量比很接近,通过对比AS 不锈钢结构优化后计算获得的晶格常数3.520Å 与试验值3.753Å[3],验证了AS 不锈钢超晶胞原子模型的可行性和可靠性。
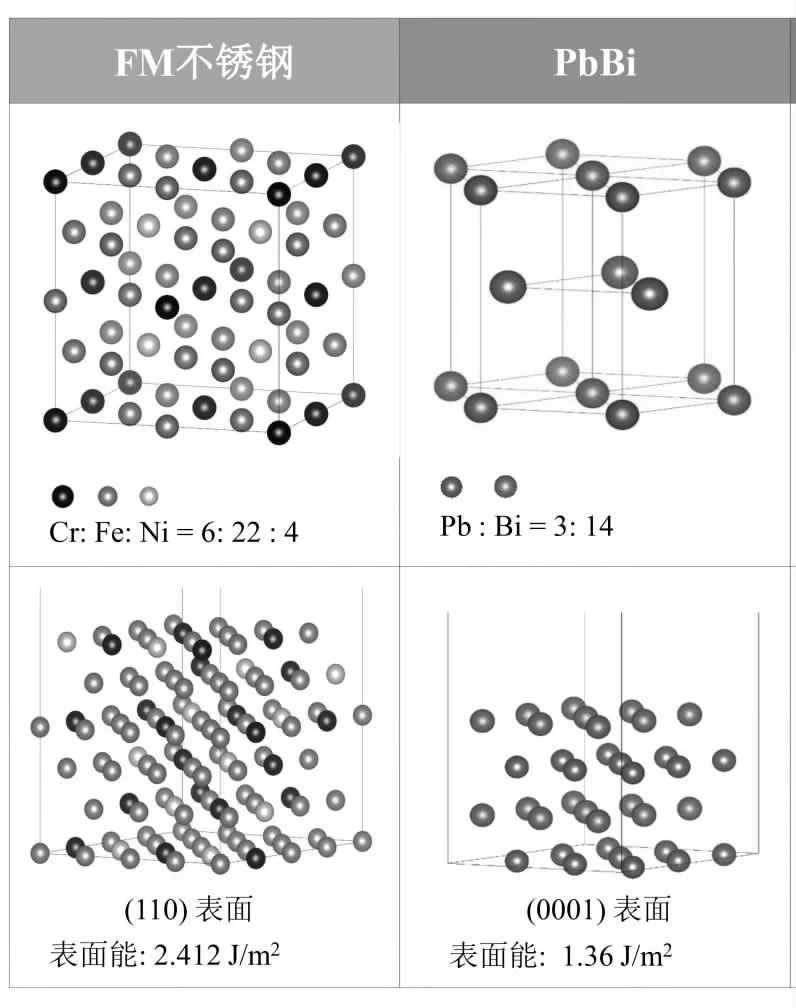
图1 AS 钢和PbBi 介质的超晶胞原子结构模型(上层)和表面原子结构模型(下层)

表1 AS 不锈钢超晶胞的点阵常数与化学成分表
PbBi 的超晶胞模型参考试验中铅铋共晶合金的组分比例,采用了原子百分比50 at. % Pb + 50 at. %Bi,折合成重量百分49.8 wt. % 的Pb 和50.2 wt. % 的Bi 构建而成,相应的晶胞参数列在表2 中。从该表中可知,本计算工作所采用的PbBi 重量百分比与实验值很接近。运用能量最低的原则,对PbBi 合金所有可能的原子结构进行了计算,优化后获得的PbBi 超晶胞结构为密排六方结构,其原子结构模型如图1 所示。优化后获得的PbBi 超晶胞的点阵常数和生成热列在表2 中,由该表可知,计算获得的PbBi 超晶胞结构的点阵常数是a=3.541 Å,c=5.841Å,与相应的实验值(a=3.5058Å,c=5.797Å)[4]符合得很好。另外,本计算工作获得的PbBi 超晶胞结构的生成热也与相应的实验值比较接近。因此,构建并优化后获得的PbBi 超晶胞原子模型是可靠的。

表2 PbBi 超晶胞的成分、点阵常数和生成热
2.3 表面原子结构模型
获得AS 不锈钢、PbBi 介质的超晶胞原子结构模型后,基于能量最低原理计算超晶胞原子结构模型中晶面的表面能,得到表面原子结构模型。对于各晶面,根据如下公式计算其表面能

其中,Ys、Es、Eb分别代表表面能、表面模型的总能和表面模型对应的体材料的能量;A 是指表面处的面积。
以AS 不锈钢的原子结构模型为基础,优化构建AS不锈钢表面原子结构模型,选取了上述AS 钢超晶胞面心立方结构的几种最常见晶面(100),(110)和(111)进行测试,经过分析对比发现(111)表面的表面能为最低。通过第一性原理对(111)晶面的原子结构、表面能、电子结构、元素偏聚等情况分别进行测试和优化,以找到AS 钢具有较低能量的表面类型。计算表明,结构优化后AS 钢的最低表面能为2.412 J/m2,其表面原子结构模型如图1所示。
PbBi 合金的密排面为(0001)表面,经过测试,PbBi 合金(0001)表面的表面能最低,图1 显示了PbBi (0001)表面的原子结构,由公式(1)计算得到PbBi (0001)表面的表面能为1.36 J/m2。
3 结果与讨论
采用第一性原理分子动力学方法模拟AS/PbBi 界面腐蚀过程中的动力学过程,获得AS/PbBi 界面界面随时间变化的原子结构、界面结合强度等,分析AS 钢铅铋腐蚀性能和微观腐蚀机理。
基于图1 中AS 不锈钢和PbBi 介质的稳定表面,构建如图2 所示两种AS/PbBi 界面原子结构模型,分别为AS/PbBi-1 和AS/PbBi-2。对AS/PbBi-1 和AS/PbBi-2 界面模型,根据下面的公式(2)和(3)分别计算界面模型的界面能(Eint)和分离功(Wsep):

图2 AS/PbBi 界面的两种界面原子结构模型以及两种AS/PbBi 界面模型中铁原子的态密度图

式中,Etot为界面的总能量,Ebulk-up和Ebulk-down分别为界面模型中上层面和下层面的能量,Eup和Edown分别为界面分离后上下两层面的能量。表3 列出了计算所获得的AS/PbBi-1 和AS/PbBi-2 界面的界面能和分离功。可以看出,AS/PbBi-2 界面模型的界面能较低,分离功较高,说明Bi 原子与AS 不锈钢形成的界面模型更稳定,结合强度更高。图2 对比了AS/PbBi-1 和AS/PbBi-2 两种界面模型中铁原子能态密度图,也可以看出AS/PbBi-2 界面峰值更为局域,其结合强度更高。

表3 AS/PbBi 界面的界面能和分离功
以结合强度更高的AS/PbBi-2 界面为研究对象,研究空位和O 原子对界面性能的影响。其中,空位及O 原子位置的选取为:对于空位,在AS 端主要考虑在界面处形成Fe 空位、Cr 空位和Ni 空位,在PbBi 端,考虑界面处形成Bi 空位;对于O 原子掺杂,主要考虑分别放置在能量较低的AS 端和PbBi 端的界面层。通过公式(2)和(3),分别计算加入空位和O 原子后AS/PbBi-2 界面的界面能和分离功,计算结果列在表4 中。

表4 含有空位和氧原子的AS/PbBi-2 界面模型的界面能和分离功
针对空位的情况,从表3 中可以看出,Fe 空位、Cr 空位、Ni 空位和Bi 空位都会提高AS/PbBi 界面的界面能,即空位降低AS/PbBi 界面的稳定性。另外,Fe 空位、Cr 空位、Ni 空位和Bi 空位都降低AS/PbBi 界面的分离功,也就是说,空位使得AS/PbBi 界面的结合强度下降,特别是Fe 空位和Ni 空位大幅度地降低了AS/PbBi 界面的结合强度。对于掺杂的氧原子,AS/PbBi 界面的界面能和分离功都有所降低。这说明,溶解氧会使AS/PbBi 界面的稳定性提高,而界面的结合强度则降低。
4 结论
本文以AS 钢为研究对象,采用第一性原理和分子动力学模拟的计算方法分别研究了AS 钢与PbBi 的相互作用及腐蚀行为,研究结果可以从原子及分子尺度揭示AS 钢在铅铋腐蚀介质中的耐腐蚀性能和微观腐蚀机制。研究结果发现,空位和溶解氧降低AS/PbBi 界面的结合强度,其中Ni 空位和Fe 空位的降低程度更显著;空位降低(Fe,Cr)3O4/PbBi 界面的稳定性和结合强度,且氧空位的降低程度更显著。
