牙鲆感染迟钝爱德华氏菌的miRNA转录组分析
2022-04-15山亚男郑津辉
山亚男,郑津辉,高 虹
(1.天津师范大学生命科学学院,天津 300387;2.天津师范大学天津市动植物抗性重点实验室,天津 300387)
牙鲆(Paralichthys olivaceus)属鲽形目(Pleuronectiformes)牙鲆科(Paralichthyidae)牙鲆属(Paralichthys),是重要的水产养殖鱼类.牙鲆生长过程中易感染细菌、病毒和真菌等,从而引发疾病,影响牙鲆的产量和品质.由于细菌感染导致的发病率较高且传染性强,比病毒和真菌的覆盖面更广泛[1-2],因此细菌感染致病是牙鲆养殖研究中需要重点解决的问题.迟钝爱德华氏菌(Edwardsiella tarda)是对水产动物有严重危害的致病菌,主要通过鱼类的消化道、鳃或受伤表皮侵入鱼体[3],目前发现该种细菌能够在二十多种鱼类中引发病害,如牙鲆、中华鳖、鲤鱼等.迟钝爱德华氏菌是一种人、鱼共患病原菌[4-5],对人类健康存在潜在威胁.全面深入研究牙鲆对迟钝爱德华氏菌免疫应答的分子机理,能够为建立一种有效的可预防和控制该病菌感染的免疫防治新方法提供理论基础.
MicroRNAs(miRNAs)是一类小的非编码RNA,通过识别同源序列和干扰转录、翻译或表观遗传过程来调控基因表达.目前对鱼类miRNA的研究相对较少,大部分miRNA的研究还是集中在疾病、常见动物或者植物方面[6-7].mi RNA在鱼类的免疫系统中有重要作用,很多差异表达miRNA参与入侵感染所产生的炎症反应,表达量被上调或下调.如Sha等[8]研究发现,半滑舌鳎被鳗弧菌侵染后,其肝脏、头肾、脾和肠中有10个差异显著表达的miRNA;Xia等[9]研究发现,尖吻鲈感染弧菌后,体内检测到了63种miRNA,其中34种表达量上升,12种表达量下降;Ordas等[10]使用沙门氏伤寒杆菌对斑马鱼胚胎进行攻毒,发现Toll样受体信号通路激活并诱导miR-146a/b表达上调.另外,聚肌苷酸胞苷酸诱导的大黄鱼、柱状黄杆菌侵染的鲤鱼、嗜水气单胞菌侵染的草鱼和虹彩病毒属肿大细胞病毒侵染的牙鲆中,也存在相似的miRNA差异性表达结果[11-13].因此,找到鱼类参与免疫应答的关键miRNA并鉴定其功能,将有助于深入理解鱼类感染病菌的生理病理活动以及深度开发和应用mi RNA.本研究以牙鲆的头肾组织为实验材料,用迟钝爱德华氏菌侵染牙鲆,利用高通量测序技术分析牙鲆感染迟钝爱德华氏菌后miRNA的差异表达情况,为进一步评估mi RNA在牙鲆免疫调控系统中的作用提供重要的理论依据.
1 材料与方法
1.1 材料及处理
实验用牙鲆购自天津市西青区某水产批发市场,从中选取9尾生长发育状态良好的个体,体长约35 cm,大小均匀.将牙鲆分为感染组和对照组,每组3尾,设置3个平行对照.选择消毒灭菌后的可控温、供氧和水过滤的水循环鱼缸,温度控制在20℃左右,盐的质量分数为1.7%~1.8%,充氧处理7 d,使牙鲆充分适应环境.牙鲆营底栖生活,不适合强光照射,因此需要对鱼缸进行遮光处理.实验开始前将牙鲆放在上述水环境中暂养1 d使其适应实验环境,每天喂养1次.
实验用迟钝爱德华氏菌由天津市水产养殖病害防治中心提供,保存在本研究实验室.将其接种于pH值为7.2的LB液体培养基中,放入28℃恒温振荡培养箱(200 r/min)中培养24 h.当菌液OD600等于1.0时,在4℃、12 000 r/min条件下离心10 min,收集菌体.感染前用PBS离心洗涤菌体3次,用血球计数板计数.从6尾牙鲆中任意选取3尾作为感染组,用PBS将菌液稀释到1×107CFU/mL,立即注射进感染组牙鲆体内.分别在感染3 h和6 h时对牙鲆进行解剖,取出头肾组织,将其剪成小块放入冷冻管中,立即放入液氮中冷冻,之后保存于-80℃冰箱中,用于RNA提取、转录组测序和生物信息学分析.委托上海元莘生物医药科技有限公司完成转录组测序.
1.2 miRNA测序结果的生物信息学分析
通过高通量测序平台(Illumina HiSeq)对牙鲆的miRNA进行测序,得到原始的FASTQ数据,里面含有带接头、低质量的序列(raw reads).为了保证后续分析的准确性,运用Fastx-Toolkit软件对raw reads进行质量控制,得到高质量的测序结果(clean reads),基于clean reads进行生物信息学分析.为了确定已知的miRNA,将得到的高质量测序结果与miRBase数据库中斑马鱼(Danio rerio)的miRNA前体及成熟体序列进行比对.
1.3 差异表达miRNA的筛选及其靶基因功能分析
对各样本的已知miRNA进行表达量统计分析,利用TPM对表达量进行均一化处理.应用DEGseq2软件对表达数据进行统计学分析,筛选不同样本间差异显著的基因.应用miRanda靶基因预测软件预测差异表达miRNA的靶基因.应用Gene Ontology、KEGG pathway软件对靶基因的代谢通路或信号转导途径进行富集分析.
1.4 差异表达miRNA的荧光定量PCR验证
为了验证本次筛选差异表达miRNA的准确性,选择6个差异表达的miRNA(miR-730、miR-727-5p、miR-127-3p、miR-205、miR-34a-5p、miR-202-3P),采用荧光定量PCR方法,分析它们在牙鲆注射迟钝爱德华氏菌后的相对表达量变化.使用HiScript 1st Strand cDNA Synthesis Kit(cDNA一链合成试剂盒)对所提取的RNA进行逆转录处理,使用Primer5设计差异表达miRNA的PCR引物,引物序列如表1所示.使用AceQ qPCR SYBR Green Master Mix(without ROX)试剂盒进行实时荧光定量PCR反应,每个样本设置3个平行管,取扩增后所得3个平行管Ct值(即每个反应管内的荧光信号到达设定的域值时所经历的反应循环数)的平均值.

表1 6个差异表达miRNA的实时定量表达引物序列Tab.1 Real time quantitative expression primer sequences of six differentially expressed miRNAs
2 结果与分析
2.1 牙鲆对照组和感染组的miRNA测序数据
通过高通量测序平台得到牙鲆感染组和对照组的转录组数据.原始数据统计:对照组有11 071 405条raw reads,感染组(3 h)有15 770 243条raw reads,感染组(6 h)有15 122 498条raw reads.对raw reads进行质量控制,得到clean reads:对照组有9 571 465条clean reads,GC含量为68.59%;感染组(3 h)有13 277 647条clean reads,GC含量为68.08%;感染组(6 h)有12 868 934条clean reads,GC含量为70.44%.不同处理组中miRNA的长度分布如图1所示.由图1可以看出,本研究中clean reads的序列长度大部分在21~23 nt,其中22 nt的比例最大,这与miRNA的普遍分布长度一致.

图1 不同处理组中miRNA长度分布Fig.1 Length distribution of miRNA in different treatment groups
2.2 牙鲆对照组和感染组的miRNA碱基分析
分析对照组和感染组中牙鲆miRNA的碱基组成:对照组中,腺嘌呤(A)、尿嘧啶(U)、胞嘧啶(C)、鸟嘌呤(G)所占比例分别为25.81%、28.42%、19.38%、26.39%;感染组(3 h)中,4种碱基所占比例分别为26.15%、28.04%、19.51%、26.30%;感染组(6 h)中,4种碱基所占比例分别为26.17%、28.12%、19.26%、26.45%.胞嘧啶在3组转录组中的比例均最低,这与房路京等[14]对大黄鱼的研究结果一致,说明不同物种之间的miRNA碱基种类所占比例存在着相似之处.
2.3 牙鲆差异表达miRNA分析
根据筛选标准|log2(foldchange)|>1和Q值<0.01,对差异表达miRNA进行评估.从感染组(3 h)中筛选得到12个差异表达miRNA,其中2个上调,10个下调;从感染组(6 h)中筛选得到32个差异表达miRNA,其中20个上调,12个下调.感染组(3 h)与感染组(6 h)的差异表达miRNA对比交集如图2所示.由图2可以看出,相较于感染3h,感染6h有更多miRNAs的表达发生了变化,说明不同的miRNAs作用时间不同,随着感染时间的延长,越来越多的miRNAs参与到免疫反应中.
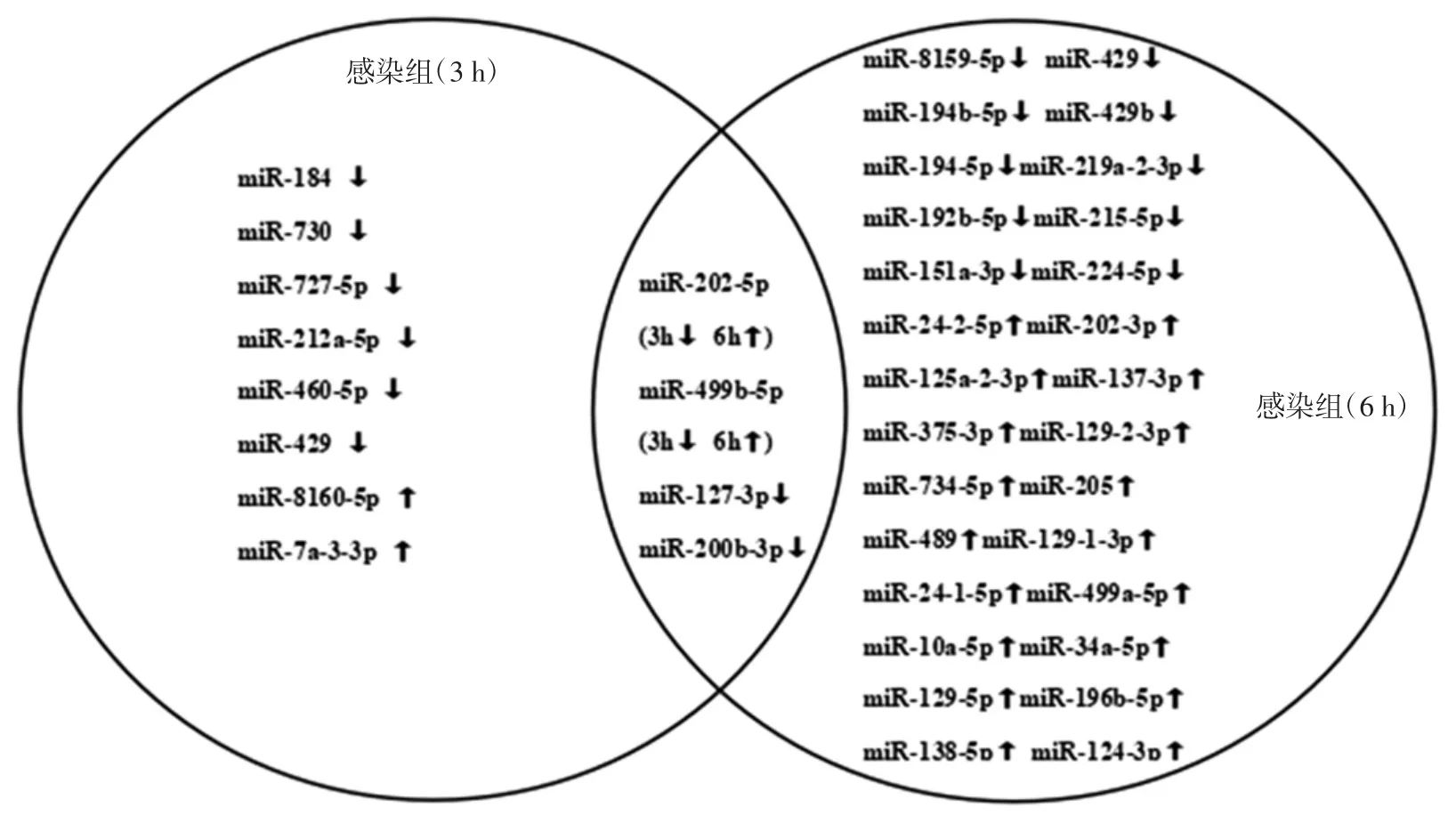
图2 感染组(3 h)和感染组(6 h)的差异表达miRNA对比交集Fig.2 Comparison of miRNA between the infection group(3 h)and the infection group(6 h)
2.4 差异表达miRNA的靶基因GO富集分析
对靶基因进行GO通路富集分析,结果显示,感染组(3 h)中有2 793条差异显著的富集通路(P<0.05),其中包括2 021个生物过程(biological process,BP)、315个细胞组分(cellular component,CC)和457个分子功能(molecular function,MF);感染组(6 h)有3 048条差异显著的富集通路(P<0.05),其中包括2 235个生物过程、342个细胞组分和471个分子功能.对比分析2个感染组,结果显示,差异显著的富集通路有2 803条(P<0.05),其中包括1 984个生物过程、341个细胞组分和478个分子功能.这些差异显著的富集通路大部分跟生物过程有关,如生长发育、信号转导、细胞通讯等.每个部分选取20个最富集的通路,做GO富集柱状图,结果如图3所示.由图3(a)可以看出,感染组(3 h)生物过程中候选靶基因富集程度最高的GO条目是生物调节(biological regulation),其次是生物过程调节(regulation of biological process);细胞组分中富集程度最高的GO条目是质膜(plasma membrane),其次是细胞外围(cell periphery);分子功能中富集程度最高的GO条目是结合(binding),其次是细胞骨架蛋白结合(cytoskeletal protein binding).由图3(b)可以看出,感染组(6 h)生物过程中候选靶基因富集程度最高的GO条目是生物调节,其次是多细胞生物过程(multicellular organismal process);细胞组分中富集程度最高的GO条目是细胞外围,其次是质膜;分子功能中富集程度最高的GO条目是结合,其次是蛋白质的结合(protein binding).由图3(c)可以看出,感染组(3 h vs 6 h)生物过程中候选靶基因富集程度最高的GO条目是发育过程(developmental process),其次是身体结构发育(anatomical structure development);细胞组分中富集程度最高的GO条目是细胞外围,其次是质膜;分子功能中富集程度最高的GO条目是结合,其次是蛋白质的结合.
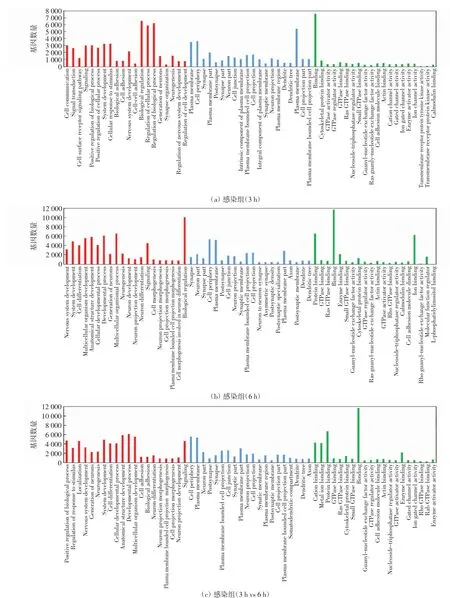
图3 GO富集柱状图Fig.3 Histogram of GO enrichment
2.5 靶基因KEGG富集分析
对牙鲆感染组差异显著表达的miRNAs进行KEGG富集分析.感染组(3 h)差异显著表达的miRNAs共获得71个KEGG富集通路(P<0.05),其中包括31个有机系统、17个环境信息处理、6个细胞过程、13个人类疾病和4个代谢通路.选取差异显著的30条通路绘制散点图,结果如图4(a)所示,富集程度最大的KEGG通路是轴突导向(axon guidance),其次是谷氨酸突触(glutamatergic synapse),富集靶基因数目最多的是丝裂原活化蛋白激酶信号通路(MAPKsignaling pathway).感染组(6 h)差异显著表达的miRNAs共获得69个KEGG富集通路(P<0.05),其中包括33个有机系统、18个环境信息处理、6个细胞过程、10个人类疾病和2个代谢通路.选取差异显著的30条通路绘制散点图,结果如图4(b)所示,富集程度最大的KEGG通路是轴突导向,其次是Rap1信号通路(Rap1 signaling pathway),富集靶基因数目最多的是癌症通路(pathways in cancer).将感染组(3 h)和感染组(6 h)两组进行对比分析,共得到79个KEGG富集通路(P<0.05),其中包括33个有机系统、19个环境信息处理、6个细胞过程、20个人类疾病和1个代谢通路.选取差异显著的30条绘制散点图,结果如图4(c)所示,富集程度最大的通路是轴突导向,其次是钙信号通路(calcium signaling pathway),富集靶基因数目最多的是丝裂原活化蛋白激酶信号通路.这些信号通路共同参与牙鲆细胞的免疫调节以及分裂分化等生物学过程.
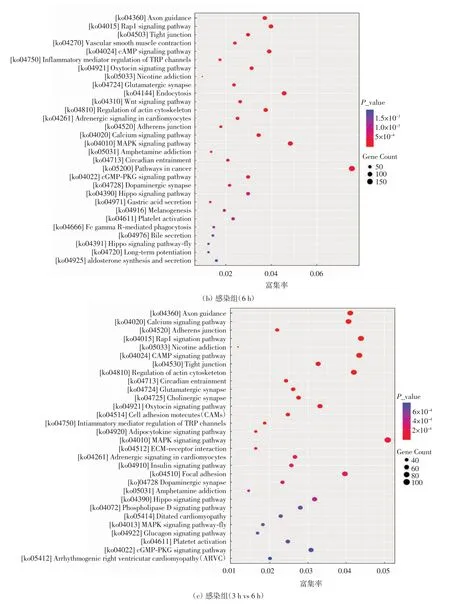
图4 KEGG富集散点图Fig.4 Catter plot of KEGG enrichment
2.6 差异表达miRNA的实时荧光定量PCR验证
为了验证高通量测序结果的可靠性,选取靶基因,对与免疫相关的6个差异表达miRNA(miR-730、miR-727-5p、miR-127-3p、miR-205、miR-34a-5p、miR-202-3p,其中3个表达上调,3个下调)进行实时荧光定量PCR验证,结果如表2所示.miR-730预测的靶基因为甘露糖受体(mannose receptor,MR),包括牙鲆阳离子非依赖性甘露糖-6-磷酸受体样、牙鲆巨噬细胞甘露糖受体1样(X1、2、3)、牙鲆甘露糖受体C型1和牙鲆胰岛素样生长因子2受体;miR-727-5p预测到的靶基因是牙鲆C型甘露糖受体2样;miR-127-3p预测到的靶基因是牙鲆C型甘露糖受体2样;miR-205预测的靶基因之一是TIRAP,这是Toll样受体(Toll-like receptor,TLR)信号通路中的接头分子,上接TLR,下接MyD88;miR-34a-5p预测的靶基因是TLR13;miR-202-3p预测的靶基因之一是TLR7.
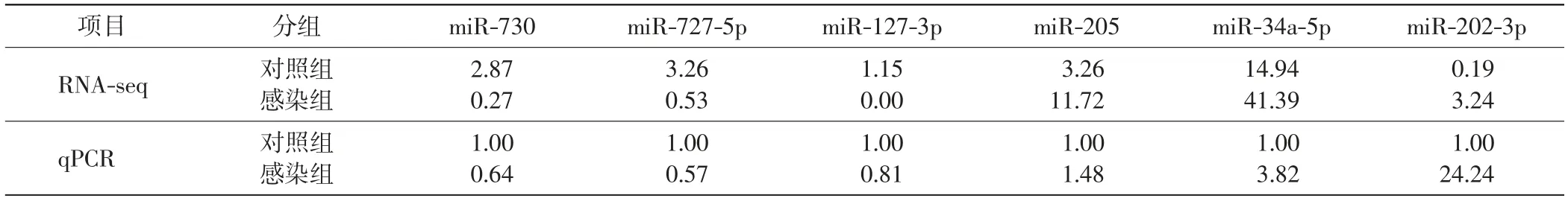
表2 实时定量PCR和转录组分析结果Tab.2 Results of qPCR and RNA-seq

由表2可知,6个miRNA的实时荧光定量PCR相对表达量结果和高通量测序结果趋势一致,miR-730、miR-727-5p、miR-127-3p表达下调,miR-205、miR-34a-5p、miR-202-3p表达上调,由此表明转录组测序结果可信.
3 讨论与结论
本研究用迟钝爱德华氏菌感染牙鲆,利用高通量测序平台对牙鲆的头肾组织进行转录组测序,并对数据进行质量控制,得到clean reads.对照组中得到9 571 465条clean reads,GC含量为68.59%;感染组(3 h)中得到13 277 647条clean reads,GC含量为68.08%;感染组(6 h)中得到12 868 934条clean reads,GC含量为70.44%.分析牙鲆对照组与感染组中miRNA的碱基组成发现,胞嘧啶所占比例均最低.利用RNA-seq测序技术及生物信息学分析,得到牙鲆感染迟钝爱德华氏菌过程中头肾组织的差异表达miRNA结果为:感染组(3 h)有12个差异表达miRNA,其中2个上调,10个下调;感染组(6 h)有32个差异表达miRNA,其中20个上调,12个下调.牙鲆被迟钝爱德华氏菌感染的时间不同,miRNA的反应程度也不同,感染6 h时应激反应处于高峰,预测miRNA可能参与了牙鲆的稳态调节.选取与模式识别受体(TLR、MR)信号通路相关的6个差异表达miRNA进行实时荧光PCR验证,所得结果与转录组测序结果一致,表明转录组测序结果可信.感染组(6 h)的3个miRNA均发生上调,这3个miRNA的靶基因与TLR信号通路有关,可能与牙鲆的免疫负调节相关,miRNA的表达上调,它对应的靶基因TLR的表达应下调.本课题组前期研究结果显示,牙鲆感染迟钝爱德华氏菌过程中,感染6 h时多数TLR基因处于表达峰值.
miRNA在抗细菌的先天免疫反应中起着非常重要的负反馈调控作用,这种调控可以确保免疫系统发生免疫反应时不会出现过激反应.TLR信号必须受到严格控制以避免产生过度炎症,并且在组织感染或损伤后允许组织修复恢复稳态.miRNA是TLR信号的重要控制因子,部分miRNA由固有免疫细胞中的TLR激活诱导,同其他miRNA一起通过靶向TLR通路中的信号分子miRNA起作用[15].TLR信号通路中存在着一些具有重要调控作用的调节因子,其调控可以是正向也可以是负向的,miRNA还可以通过作用于这些调节因子间接发挥对TLR信号通路的调控作用.miRNA被证明是固有免疫和适应性免疫之间的重要连接,它们的失调可能在炎症疾病的发病机制中起到重要作用[16-22].本研究发现,差异表达miRNA对应的靶基因所参与的信号通路大部分跟生物过程以及细胞组分中的膜成分有关,如生长发育、信号转导、细胞通讯、质膜等.生物过程中候选靶基因富集程度最高的GO条目是生物调节,富集程度高的通路大多跟免疫调节以及分裂分化等生物学过程相关联,质膜具有识别和传递信息的功能,以上结果可以为牙鲆免疫基因的筛选和免疫系统的研究提供理论依据.这些差异表达miRNA及其靶基因可能与牙鲆抗迟钝爱德华氏菌感染过程密切相关,对研究牙鲆的免疫调控机制具有重要指导意义,其所提供的差异表达量变化的信息可以为后续研究牙鲆应对迟钝爱德华氏菌刺激时靶基因的筛选以及靶基因所起作用提供理论依据.
