蒽二噻吩分子连接铁磁锯齿边碳化硅纳米带的巨幅度自旋整流*
2022-04-15李佳锦刘乾伍丹邓小清张振华范志强
李佳锦 刘乾 伍丹 邓小清 张振华 范志强
(长沙理工大学物理与电子科学学院,柔性电子材料基因工程湖南省重点实验室,长沙 410114)
利用非平衡格林函数结合密度泛函理论,研究了顺式蒽二噻吩和反式蒽二噻吩分子连接锯齿边碳化硅纳米带的自旋输运特性,并在铁磁场下观察到自旋向上和自旋向下具有同方向的自旋整流特性.在铁磁场下,边缘碳原子或者硅原子双氢原子钝化可以改变锯齿边碳化硅纳米带的本征金属性,使其转变为半导体.顺式蒽二噻吩器件和反式蒽二噻吩器件的自旋向上电流-电压特性可以呈现显著的自旋整流效应,相应的最大自旋整流比分别接近1011和1010.此外,由于自旋向上和自旋向下电流值之间的巨大差异,两个器件的电流-电压特性都在正偏压区域呈现出完美的自旋过滤行为.以上发现对未来设计自旋功能分子器件具有重要意义.
1 引言
随着硅基半导体器件尺寸缩小到纳米级,显著的小尺寸效应和量子效应使其难以在著名的集成电路摩尔定律下继续延伸和推进[1].全世界的科学家都在寻找体积更小、功能更好的设备来解决这些问题.其中,分子器件和分子电子学为电子器件的进一步小型化提供了机会[2-4].迄今为止,科学家们在对分子热电和光电器件的输运行为进行实验和理论研究时,观察到了一系列优良特性[5-9],如负微分电阻[10,11]、分子二极管[12,13]、分子开关[14,15]、分子马达[16]、分子场效应晶管[17]等.虽然分子器件比传统的硅基半导体器件小,但信息传输的载体仍然是电子.20 世纪80 年代,法国科学家在外磁场作用下观察到Fe/Cr 周期性多层膜中电阻变化率高达50%的巨磁阻效应,电阻变化率高达50%,标志着自旋电子学的诞生.1995 年,科学家在Fe/Al2O3/Fe 夹层结构中观察到一个大的隧穿磁阻,这为自旋电子学开辟了另一个新方向.自旋电子学以电子自旋为基础,用于信息传输、处理和存储,其具有传统半导体电子器件无法比拟的优势,如运行速度更快、集成度更高、能耗更低等,因此成为近年来的研究热点[18-20].
近年来,以石墨烯为代表的二维材料由于其优异的物理和化学性能而成为全球研究热点[21-25].目前,实验成功制备的本征二维材料普遍不具有磁性,这极大地限制了二维材料在自旋电子学领域的应用.然而,当二维材料被切割成纳米带时,如石墨烯纳米带[26]、硅烯纳米带[27]、碳化硅纳米带[28]、磷烯纳米带和二硫化钼纳米带[29,30],由于边缘状态的存在,它们可以显示磁性.同时,以上述纳米带作为电极连接单个有机分子的磁性输运器件也表现出多种自旋电子输运特性,如自旋极化、自旋过滤、自旋开关、自旋整流、巨磁阻等效应[31-36].其中,自旋整流效应是指磁性器件的自旋电流仅在正向偏压下导通,在反向偏压下关闭的单向导电特性.它在开关电路、稳压电路、限幅电路和电容电路中都有重要的应用.目前,能够实现自旋电流整流的纳米器件或分子器件通常需要在器件电极处加上反铁磁场,这无疑增大了实现的复杂性和难度[37-40].另外,两种自旋流整流的单向传导方向相反,整流比一般不高.因此,如何在铁磁场存在的情况下实现高整流比的自旋电流整流将是一个非常有意义的研究课题.本文研究了不同边缘双氢原子钝化锯齿边碳化硅纳米带连接蒽二噻吩(anthradithiophene,ADT) 分子的自旋输运特性.特别是,蒽二噻吩与并五苯是等电子的,并五苯是有机场效应晶体管中研究最多的有机半导体材料[41,42].然而,关于蒽二噻吩分子的自旋输运特性还没有详细的报道.研究发现顺式蒽二噻吩器件(cis-ADT)和反式蒽二噻吩(trans-ADT)器件的自旋向上电流电压特性在铁磁场下可以呈现显著的自旋整流特性,对应的自旋电流整流比分别接近1011和1010.
2 模型与方法
图1 为不同边缘双氢原子钝化锯齿边碳化硅纳米带连接蒽二噻吩分子的器件几何结构.与石墨烯纳米带一样,二维碳化硅也可裁剪成锯齿边碳化硅纳米带和扶手椅边碳化硅纳米带.锯齿边碳化硅纳米带呈现金属性,而扶手椅边碳化硅纳米带是带隙巨大的半导体.即使在外磁场作用下,扶手椅边碳化硅纳米带的自旋能带结构显示其两种自旋态的带隙仍然很大,无法作为自旋器件的理想电极.为了处理实际器件的无限边界问题,将其分为3 个区域:左电极、右电极和中心散射区.左右电极分别是底部边缘硅原子和顶部边缘碳原子双氢原子钝化的锯齿边碳化硅纳米带.中心散射区的分子是蒽二噻吩的两种异构体,顺式蒽二噻吩和反式蒽二噻吩,它们是具有两个末端苯基团被噻吩环取代的五苯类似物[41-43].蒽二噻吩末端的碳原子可以直接与碳化硅上的碳原子和硅原子形成共价键,从而将蒽二噻吩分子稳定地锚接在碳化硅上构成分子器件.图上明确标记了硫原子在顺式蒽二噻吩和反式蒽二噻吩中的位置.由于异构体分离困难,蒽二噻吩最初被合成反式异构体和顺式异构体的混合物.此外,两种异构体的结构变化会导致一些电荷重排,这也会改变两种异构体中蒽二噻吩分子器件的输运性质.因此,如果想系统地研究锯齿边碳化硅纳米带连接蒽二噻吩分子的自旋输运特性,应该考虑它的两种异构体.为简单起见,这两种不同的设备分别命名为M1和M2.在铁磁场下计算了自旋能带结构和传输特性.图1 还显示了自旋向上和自旋向下状态之间的自旋电荷密度差异的等值面图(∇ρρup-ρdown),其中ρup和ρdown分别表示自旋向上(红色)和自旋向下(蓝色)的电子密度.两个器件上左电极的碳原子和底部硅原子边缘的两个钝化氢原子上分布着自旋向上净电荷,分布在硅原子上的自旋向下净电荷非常小.两个器件上右电极的硅原子和顶部碳原子边缘的两个钝化氢原子上同样分布着自旋向上净电荷,而自旋向下净电荷分布在碳原子上.两个器件上中间蒽二噻吩上自旋向上净电荷和自旋向下净电荷的分布都非常小,并且顺式或反式异构体也不同.
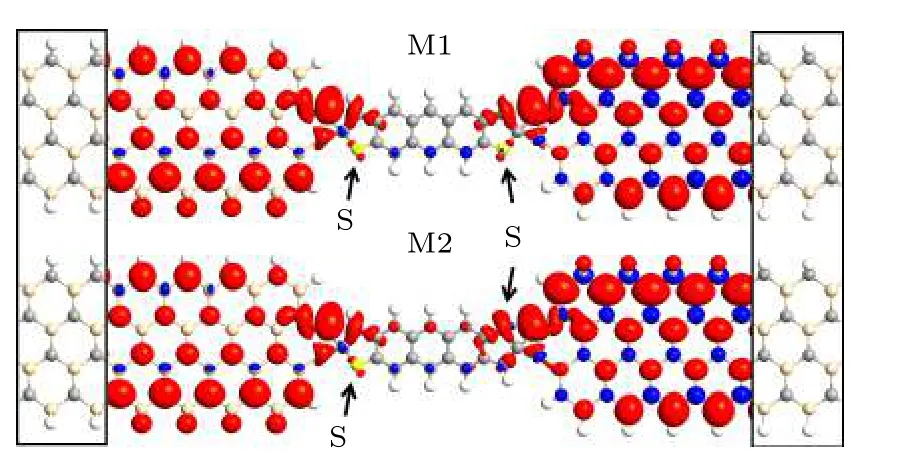
图1 两种器件的几何结构示意图.左、右黑色实心线框分别是左、右电极,为不同边缘双氢原子钝化锯齿边碳化硅纳米带的两个晶胞;自旋向上和自旋向下状态的自旋极化电荷密度差的等值图,等值均设置为0.05|e| Å—3 (1 Å=0.1 nm)Fig.1.Schematic geometric structures of two devices.The left and right black solid wire frames are the left and the right electrode,which are two unit cells of zigzag SiCnanoribbon under different edge dual-hydrogenation.Isosurface plots of the spin-polarized charge density difference of upspin and down-spin states with the isovalues all setting at 0.05|e| Å—3 (1 Å=0.1 nm).
本文中器件的几何结构优化、电子结构和自旋输运特性的计算全部由基于非平衡格林函数和密度泛函理论相结合的第一性原理计算方法 (QuantumAT 软件) 完成[44].在自旋相关广义梯度近似下,用Perdew-Burke-Ernzerhof (PBE)泛函描述了交换和关联.哈密顿量和电子密度是在能量截止值为150 Ry 的真实空间网格中计算的.用于计算块体电子结构和器件电子输运特性的k点采样在x,y和z方向分别为1×1×21和1×1×100,其中z方向是纳米带的周期方向.在x和y方向上设置至少15 Å的真空缓冲空间,以避免相互作用.器件的自旋电流由著名的朗道公式计算得到:Iσ(Vb)Tσ(E,Vb)[fL(E,Vb)-fR(E,Vb)]dE[45].其中,Tσ(E,Vb)是与自旋相关的传输系数,σ分别表示自旋向上态和自旋向下态,μL/R是左右电极的电化学势,fL(E,Vb) 和fR(E,Vb) 是左右电极的费米-狄拉克分布函数.
3 结果与讨论
图2 所示为底部边缘硅原子上双氢原子钝化的锯齿边碳化硅纳米带和顶部边缘碳原子上双氢原子钝化的锯齿边碳化硅纳米带的几何结构和自旋能带结构.锯齿边碳化硅纳米带边缘原子上的双氢原子钝化几乎不影响其平面几何结构.锯齿边碳化硅纳米带中碳氢原子之间和硅氢原子之间的键长d1和d3分别为1.09 Å和1.48 Å.但是在双氢原子钝化位置,碳氢原子之间和硅氢原子之间的键长d2和d4分别增至1.11 Å和1.52 Å,对应角度θ1和θ2分别为104.6°和103.8°.然而,双氢原子钝化可以导致电荷分布重构,这将直接影响锯齿边碳化硅纳米带的电子结构.上文计算结果表明,在铁磁场下可以观察到锯齿边碳化硅纳米带呈现本征金属性[46-50].但是计算的自旋能带结构表明,在铁磁场下,边缘碳或硅原子上的双氢原子钝化都可以将锯齿边碳化硅纳米带的本征金属性转变为半导体.在图2(a)中,自旋向上电子有很大的带隙,而自旋向下电子具有很小的带隙.自旋向上电子的价带顶接近费米能级,而导带底远离费米能级.相比之下,图2(b)中情况与图2(a)相反.自旋向上电子的带隙很小,而自旋向下电子的带隙很大.自旋向下电子的导带底接近费米能级,而价带顶远离费米能级.值得注意的是,关于边缘碳或硅原子上双氢原子钝化的锯齿边碳化硅纳米带的自旋能带计算结果与基于Heyd-Scuseria-Ernzerhof 杂化密度泛函的理论研究结果一致[46].
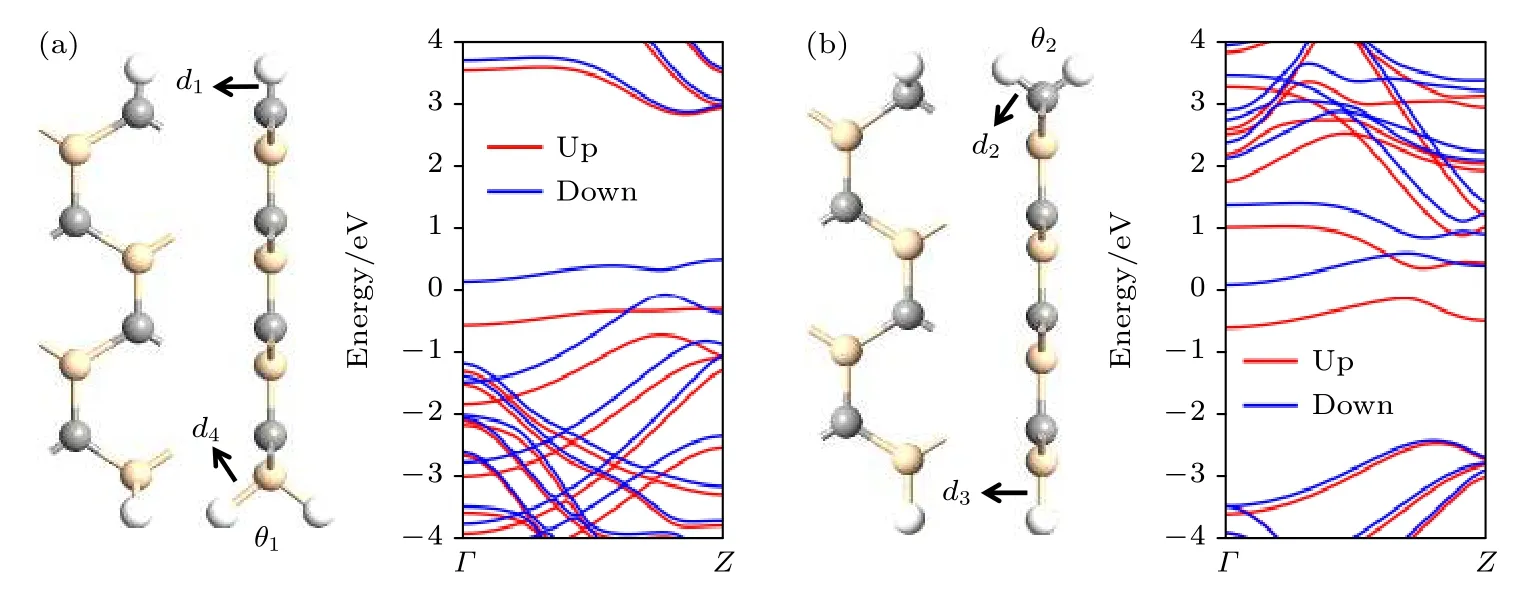
图2 (a)底部边缘硅原子双氢原子钝化的锯齿边碳化硅纳米和(b)顶部边缘碳原子双氢原子钝化的锯齿边碳化硅纳米的几何结构与自旋能带结构Fig.2.Geometric structures and spin-resolved band structures of (a) the zigzag SiC nanoribbon with the dual-hydrogenation on the silicon atom of the bottom edge and (b) the zigzag SiC nanoribbon with the dual-hydrogenation on the carbon atom of the top edge.
图3 分别显示了M1和M2 在零偏压下的自旋输运谱及透射峰值处的局域态密度空间分布.器件M1 有一个自旋向上输运峰位于—0.4 eV和一个自旋向下输运峰位于0.42 eV.自旋向上输运峰值处的局域态密度分布在左电极和中心顺式蒽二噻吩分子,在右侧电极上出现了部分局域的情况.然而,自旋向下输运峰值处的局域态密度仅仅分布于右侧电极上.中心顺式蒽二噻吩分子和左侧电极对器件态密度的贡献非常小.该结果与图1 中自旋向上和自旋向下状态之间自旋电荷密度差异等值面图完全一致.结果表明,自旋向上输运峰的系数大于自旋向下输运峰.换句话说,器件M1 的自旋向上电子传输能力高于自旋向下电子.当中心分子从顺式蒽二噻吩变为反式蒽二噻吩时,M2 的自旋向上和自旋向下输运峰仍分别位于—0.4 eV和0.42 eV.局域态密度在自旋向上输运峰的空间分布在左电极上仍然呈现有很好的离域性.但是,中心反式蒽二噻吩分子和右电极对M2 的态密度贡献已显著减弱.因此,器件M2 的自旋向上输运峰系数低于器件M1.然而,中心反式蒽二噻吩分子上自旋向下输运峰处的局域态密度空间分布有所扩大.因此,器件M2 的自旋向下输运峰系数相比器件M1略有增大.
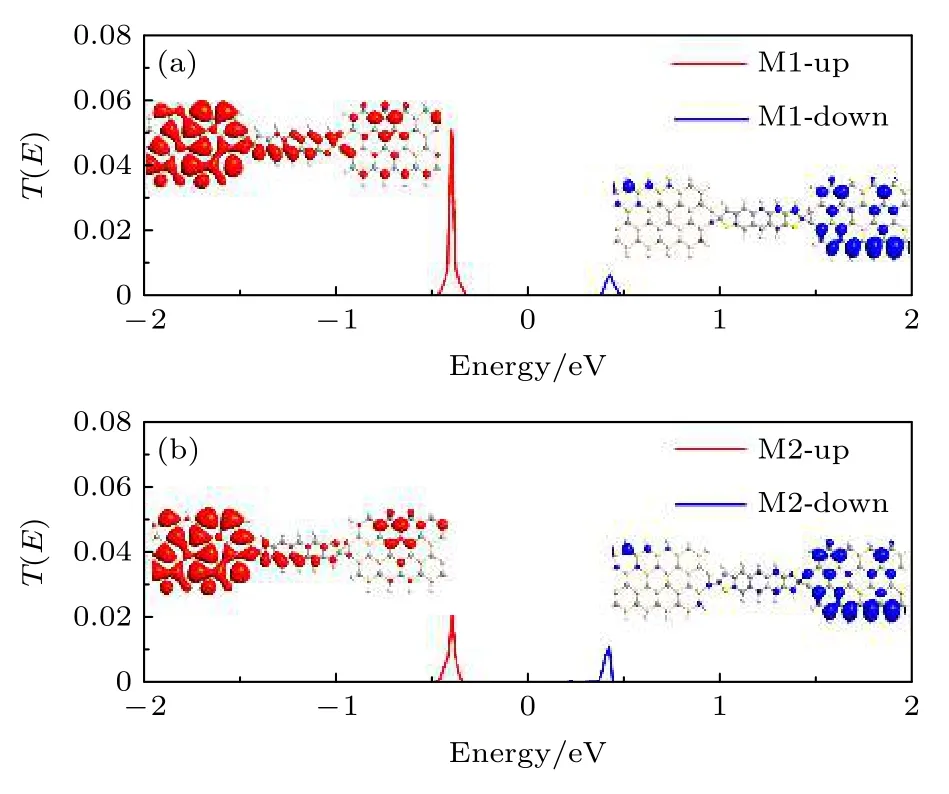
图3 零偏压下的自旋输运谱和输运峰值处的自旋向上(红色)和自旋向下(蓝色)局域态密度空间分布 (a) M1;(b) M2Fig.3.The spin-resolved transmission spectra and the spatial distributions of the up-spin (red) and down-spin(blue)local density of state at the transmission peaks under zero bias:(a)M1;(b)M2 .
图4 所示为零偏压下左电极、右电极和中心蒽二噻吩的投影态密度.分子与左电极和右电极的耦合决定了器件的输运谱[51].在图4(a)中,左电极(底部边缘硅原子上双氢原子钝化的锯齿边碳化硅纳米带)的自旋向上投影态密度在—0.4—1.5 eV 的能量范围内为零.右电极(顶部边缘碳原子上双氢原子钝化的锯齿边碳化硅纳米带)的自旋向上投影态密度在—1.5——0.62 eV 的能量范围内为零.在费米能级两侧,中心顺式蒽二噻吩有3 个自旋向上投影态密度峰,分别位于—0.4 eV,—0.32 eV和0.76 eV.然而,只有位于—0.4 eV 的中心蒽二噻吩的投影态密度峰可以同时与左右电极的投影态密度峰匹配.因此,图3(a)中器件M1 只有一个自旋向上输运峰位于—0.4 eV 处.当中心分子从顺式蒽二噻吩变为反式蒽二噻吩时,中心反式蒽二噻吩的3 个自旋向上投影态密度峰分别位于费米能级两侧的—0.4 eV,—0.24 eV和0.7 eV.但是只有位于—0.4 eV的中心反式蒽二噻吩的投影态密度峰能同时与左右电极的投影态密度匹配.此外,该投影态密度峰的系数小于顺式蒽二噻吩的输运系数.因此,图3(b)中器件M2 只有一个自旋向上输运峰也位于—0.4 eV,但是系数较小.在图4(b)中,左电极的自旋向下投影态密度在0.42—1.5 eV 的能量区域内为零.右电极的自旋向下投影态密度在—1.5—0.06 eV 的能量范围内为零.尽管中心顺式蒽二噻吩和反式蒽二噻吩在费米能级的两侧都有几个自旋向下投影态密度峰,但只有0.42 eV 的投影态密度峰可以同时与左右电极的投影态密度峰匹配.因此,图3 中M1或 M2 只有一个自旋向下输运峰位于0.42 eV.由于反式蒽二噻吩在0.42 eV 的投影态密度峰系数略高于顺式蒽二噻吩在0.42 eV 时的投影态密度峰系数,因此M2 的自旋向下输运峰系数比M1 略有增大.
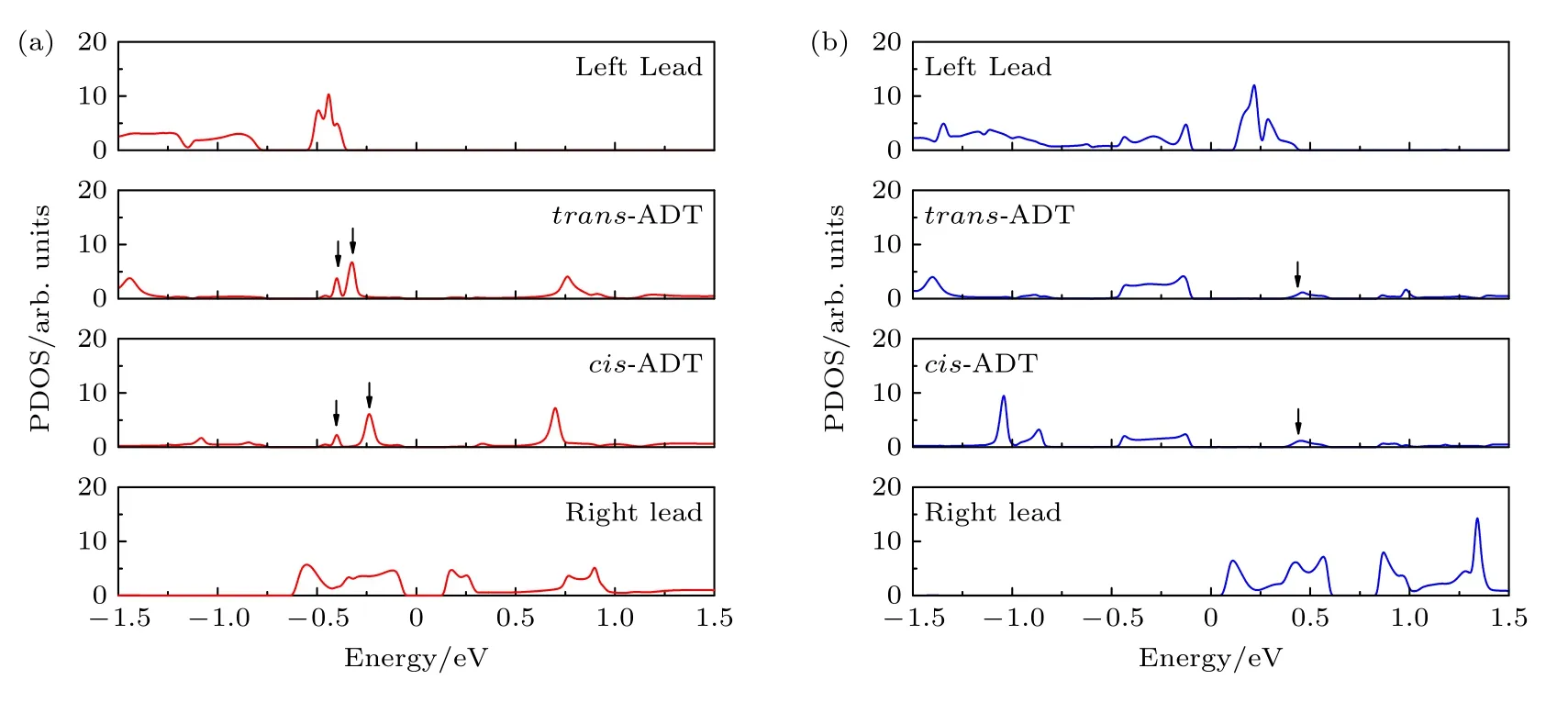
图4 (a) 零偏压下左、右电极和中央蒽二噻吩的自旋向上投影态密度;(b) 零偏压下左、右电极和中心蒽二噻吩的自旋向下投影态密度.Fig.4.(a) The up-spinprojecteddensity of states of left electrode,right electrode and central anthradithiophene at zero bias voltage;(b) the down-spinprojecteddensity of states of left electrode,right electrode and central anthradithiophene at zero bias voltage.
两种器件的自旋电流电压特性可以通过朗道公式计算得到,如图5 所示.其中正负电压的电流值均取绝对值,以方便用对数图进行比较.在图5(a)中,自旋向上和自旋向下电流几乎不随正电压的增大而变化,并且始终保持很小的值.然而,自旋向上电流在—0.15 V 后随负偏压的增大而增大,在—0.25 V 时接近8 nA.在负偏压下自旋向下电流的增大不明显.因此,当两个电极处于铁磁状态时,M1 的自旋向上和自旋向下电流电压特性都可呈现自旋电流整流效应.图5(b)显示器件M1 的自旋向下电流实际上随着正偏置电压的增大而缓慢上升.因此,M1 的对数形式的电流电压特性可以更清楚呈现自旋电流整流效应.同时可清晰看到器件M1 的自旋向上电流随着正电压的增大而迅速减小.因此可以推断自旋向上电流电压特性与自旋向下电流电压特性相比将呈现更显著的整流效应.此外,由于自旋向上和自旋向下电流值在正偏压区间的巨大差异,器件M1 的电流电压特性在正偏压区也可以表现出自旋过滤行为.
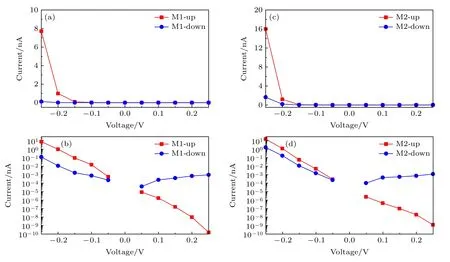
图5 器件M1 的(a)线性和(b)对数自旋电流电压特性;器件M2 的(c)线性和(d)对数自旋电流电压特性Fig.5.Spin-resolved current-voltage characteristic of M1 in (a) linear and (b) logarithmic forms;current-voltage characteristic of M2 in (c) linear and (d) logarithmic forms.
图5(c)显示,当中心分子由顺式蒽二噻吩变为反式蒽二噻吩时,电极处于铁磁状态时器件M2 的电流电压特性也会呈现自旋电流整流效应.自旋向上和自旋向下电流几乎不随正偏压的增大而变化,始终保持很小的值.然而,自旋向上电流会在—0.15 V 后随着负偏压而增大,并在—0.25 V时接近16 nA.此外,自旋向下电流会在—0.2 V 后随着负偏压而增大.图5(d)中器件M2 对数形式的电流电压特性表明自旋向上电流同样也随着正电压的增大而迅速减小.因此器件M2 的自旋向上电流电压特性与自旋向下电流电压特性相比也将呈现更显著的整流效应.此外,与M1 相比,M2 在正偏压范围内的自旋向上和自旋向下电流值差异更明显,尤其是在低偏压下.因此,器件M2 的电流电压特性在正偏压下可以表现出更完美的自旋过滤行为.为量化自旋电流整流和自旋过滤行为的水平,计算了器件的自旋电流整流比为RR=I(—V)/I(+V)和自旋过滤效率为 SFE=(Iup—Idown)/(Iup+Idown)×100%,如图6所示[52,53].从图6(a)和6(b)可以看出,随着电压从—0.05——0.25 V的变化,两种器件的自旋向上和自旋向下电流整流比都逐渐增大.如图6(a)所示,自旋向上电流整流比远大于自旋向下电流整流比.M1 的最大自旋向上电流整流比在—0.25 V 时可以接近1011,而M1的最大自旋向下电流整流比在—0.25 V 时接近102.在图6(b)中,自旋向上电流整流比也远大于自旋向下电流整流比.M2 的最大自旋向上电流整流比在—0.25 V 时可以超过1010,略低于M1.但是M2的最大自旋向下电流整流比在—0.25 V 时可以超过103,比M1 大一个数量级.两种器件的自旋向上电流整流比相比以往器件在反铁磁状态下得到的自旋电流整流要更显著.从图6(c)和(d)可以看出,两个器件的自旋过滤效率都随着偏置电压从±0.05—±0.25 V 的变化而增大,并且两个器件在正电压区域的自旋过滤效率要远远大于器件在负电压区域的自旋过滤效率.在图6(c)中,M1 在—0.05 V 时的自旋过滤效率仅为42%,在—0.1 V后超过90%.M1 在0.05 V 时的自旋过滤效率仅为—65%,然后在0.15 V 后超过—99%.在图6(d)中,M2 在负偏置电压区域的自旋过滤效率与M1 相比大大降低.M2 在—0.05 V 时的自旋过滤效率仅为10%,在—0.25 V 时的最大自旋过滤效率也仅为81%.然而,与M1 相比,M2 在正偏压区的自旋过滤效率却大大提高.M2 在 0.05 V 时的自旋过滤效率就可以达到—95%,然后在 0.1 V 后接近—100%直至0.25 V.
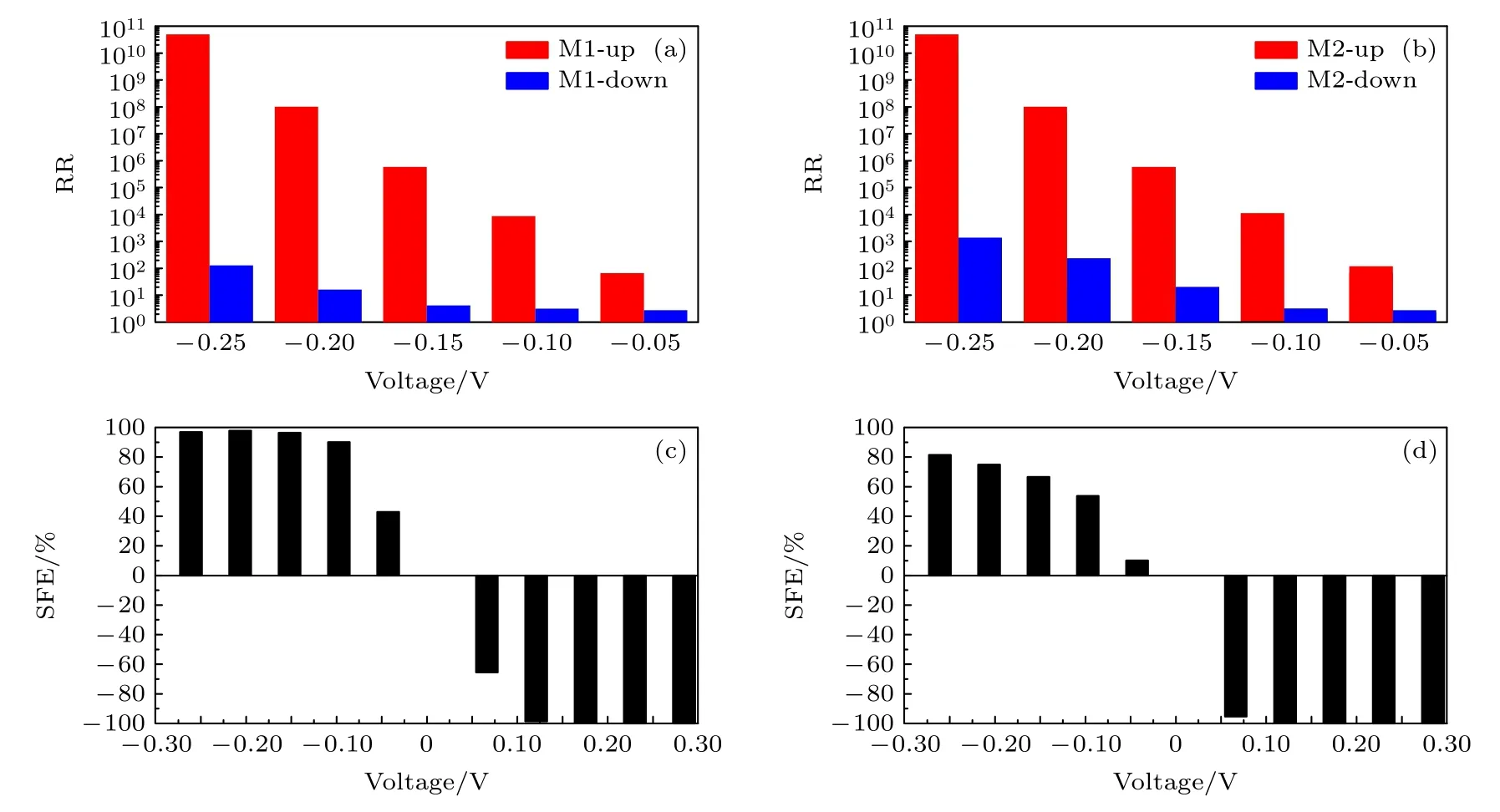
图6 (a) M1和(b) M2 的自旋向上和自旋向下电流整流比;(c) M1和(d) M2 的自旋过滤效率Fig.6.The up-spin and down-spin current rectification ratios of (a) M1 and (b) M2;the spin filtering efficienciesof (c) M1 and (d) M2.
自旋向上和自旋向下电流整流之间的巨大差异可以通过随电压演变的输运谱来解释.因此,图7给出了器件M1 在不同电压下的自旋向上和自旋向下对数输运谱比较.当对器件施加电压后,左右电极化学势分别由μL(Vb)μL(0)-eVb/2 和μR(Vb)μR(0)+eVb/2确定,因此左右电极化学势差等于eVb.在正偏压下,左电极的电势降低,右电极的电势升高.相反,在负偏压下,左电极的电势升高,而右电极的电势降低.在图7(a)中,M1 在零偏压下的自旋向上输运峰位于费米能级的左侧.随着负偏压的增大,自旋向上输运谱将向右移动,导致输运峰逐渐接近费米能级.更重要的是,由于两个电极的投影态密度匹配,输运峰系数也随着负偏压的增大而增大.最大输运系数在—0.15 V 后接近1.因此,自旋向上电流随着负偏压的增大而迅速增大.然而,M1 的自旋向上输运谱随着正偏压的增大会向左移动,导致输运峰逐渐远离费米能级.此外,由于两个电极的投影态密度不匹配,输运峰值的系数随着正电压的增大而明显下降.因此,自旋向上电流随着正偏置电压的增大而迅速减小.基于这些分析,自旋向上电流电压特性呈现出具有巨大自旋电流整流比的显著整流效应.
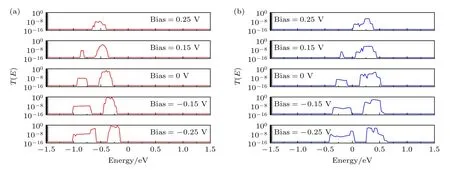
图7 M1 在不同电压下的(a)自旋向上和(b)自旋向下对数输运谱Fig.7.The up-spin (a) and down-spin (b) logarithmic transmission spectra of M1 under the different bias voltages.
在图7(b)中,M1 零偏压下主要的自旋向下输运峰值位于费米能级的右侧.此外,另一个系数很小的自旋向下输运峰位于费米能级的左侧.随着负偏压的增大,自旋向下输运谱向右移动,导致自旋向下输运主峰逐渐远离费米能级.幸运的是,随着负偏压的增大,费米能级左边的输运峰逐渐向费米能级移动,相应的系数也随之增大,导致自旋向下电流增大.施加正偏压后,M1 的自旋向下输运谱也将逐渐向右移动,导致右侧主自旋向下输运峰向费米能级移动.然而,相应的系数随着正偏置电压的增大而减小.尽管自旋向下电流在正偏压下也增大,但仍远远低于负偏压时的数值.因此,自旋向下电流电压特性也表现出自旋电流整流效应,但相应的整流比远小于自旋向上电流电压特性的自旋电流整流比.
4 结论
蒽二噻吩与并五苯都是目前研究广泛的有机半导体材料.并五苯是全碳原子构成的对称结构,而蒽二噻吩具有因硫原子位置不同而导致的顺式和反式结构.因此蒽二噻吩与并五苯相比更具有可调控的电子结构.本文研究了顺式蒽二噻吩和反式蒽二噻吩分子连接锯齿边碳化硅纳米带的自旋输运特性,并在铁磁场下观察到自旋向上和自旋向下具有同方向的自旋整流特性.研究发现边缘原子上的双氢原子钝化几乎不影响锯齿边碳化硅纳米带的平面几何结构,但可以在铁磁场下将其从本征金属性改变为半导体性.由于蒽二噻吩分子两种自旋态投影态密度与左右电极自旋态投影态密度匹配情况存在差异,顺式蒽二噻吩器件和反式蒽二噻吩器件的自旋向上电流电压特性可以呈现显著的整流效应,相应的自旋电流最大整流比分别接近1011和1010.同时,顺式蒽二噻吩和反式蒽二噻吩器件的自旋向下电流电压特性也表现出同方向的整流效应,但相应的自旋电流整流比远小于自旋向上电流电压特性.随着正电压的增大,两个器件的向下自旋电流缓慢上升.但是两个器件的自旋向上电流迅速下降.因此,自旋向上和自旋向下电流值之间的巨大差异导致两个器件在正偏压区域呈现完美的自旋过滤行为.以上发现对未来设计自旋功能分子器件具有重要意义.
