SAPO-34提升铁钒基催化剂抗碱性能研究
2022-04-14胡方方蔡思翔马艳平
胡方方,李 顺,蔡思翔,姜 宏,马艳平
(海南大学材料科学与工程学院,海南省特种玻璃国家重点实验室,南海海洋资源利用国家重点实验室,海口 570228)
0 引 言
氮氧化物(NOx)作为大气主要污染物之一,对全球环境和人类健康造成较大威胁,氨气选择性催化还原技术(NH3-SCR)是最经济有效的氮氧化物脱除技术[1]。目前,商用的V2O5-WO2/TiO2催化剂存在操作温度高、活性温度窗口窄、脱硝性能易受碱金属影响等问题[2-3],因此开发新型高效抗碱金属中毒催化剂成为重点。钒酸盐相较于传统钒氧化物,具有较高的分解温度,能够规避高温下钒挥发带来的环境污染问题。在众多的金属钒酸盐中,钒酸铁因其优异的低温催化活性、宽工作温度窗口,受到研究者的关注[4]。
在玻璃熔窑、垃圾焚烧等工作环境中所排放出的烟气中往往含有大量碱金属与碱土金属元素,极易导致催化剂中毒失活,其中碱金属的影响更为巨大[5-6]。因此,研究SCR催化剂的抗碱中毒机制具有重要意义。仲兆平等[7]通过研究钾、钠、锌、磷四种物质对SCR脱硝反应的影响,发现钾盐对于V2O5/TiO2催化剂的毒化作用最强。高凤雨等[8]通过水洗和酸洗两种方法对失活催化剂进行再生模拟,结果显示碱金属引起催化剂的中毒分为物理中毒和化学中毒,其中引起物理中毒是因为钾盐颗粒沉积对孔道造成堵塞,化学中毒是因为碱金属与催化剂表面的B酸位点反应生成V—ONa,改变了催化剂表面金属氧化物的化学环境。Gao等[9]对中毒后的催化剂进行分析,表明Na2O和K2O通过降低催化剂表面化学吸附氧含量,来抑制催化剂的脱硝活性。为了抵抗碱金属对催化剂的毒害作用,研究者们提出了一些解决方案:(1)增加载体酸性,例如徐托雨等[10]通过用硫酸化处理TiO2载体,使CeO2/TiO2催化剂具有更多的酸性点;(2)添加助剂,例如Peng等[11]通过引入Ce作为助剂提升催化剂的抗碱能力;(3)分隔活性位点,例如Huang等[12]制备锰钡矿结构(HMO)催化剂,发现其活性不受碱金属负载量的影响,其原因是催化剂活性位点位于HMO纳米棒外表面,而碱金属毒化位点位于纳米管壁内,隔绝碱金属对活性组分的毒害。
基于上述思路,本文从优化催化剂表面酸位分布、构筑抗碱中心、改善催化剂结构性质的角度出发,利用SAPO-34分子筛替代传统TiO2载体[13-15],采用经过优化的钒铁比[16],制备了Fe-VOx/SAPO-34催化剂,其优势在于:SAPO-34内部特定的孔道结构及稳定的骨架能够促进表面活性物种的均匀分散,降低碱金属对表面活性中心的物理覆盖作用;同时其表面丰富的酸位点能够作为碱金属捕获位,保护催化剂表面的酸性位点与活性中心,使得催化剂的吸附-反应过程能够延续。在此基础上,通过一系列表征手段阐明Fe-VOx/SAPO-34与Fe-VOx/TiO2的结构性质、物化性质以及催化反应路径上的区别,分析该催化剂具有良好碱金属耐受性的深层原因。
1 实 验
1.1 试剂与材料
Fe(NO3)3·9H2O、NH4VO3、草酸、尿素、纳米TiO2均购于国药集团化学试剂有限公司,纯度为分析纯。SAPO-34分子筛购于南开大学催化厂(n(P2O5)∶n(SiO2)∶n(Al2O3)=1∶1∶1)。
1.2 催化剂的制备
采用尿素沉淀法制备Fe-VOx(n(Fe)∶n(V)=3∶1)[16]:称取12.12 g的Fe(NO3)3·9H2O超声溶解于200 mL去离子水中得到溶液A。称取3 g的草酸加入50 mL去离子水中,待草酸完全溶解后加入2.34 g NH4VO3得到溶液B。将溶液A缓慢滴加于溶液B充分溶解后,加入36 g尿素。将混合溶液在90 ℃下油浴8 h,反应结束后,待溶液冷却至室温,过滤离心、干燥、在450 ℃条件下煅烧3 h得到Fe-VOx样品。
采用浸渍法制备Fe-VOx/SAPO-34:称取0.15 g的Fe-VOx充分溶解于50 mL去离子水中,加入1 g SAPO-34分子筛,充分搅拌后,旋转蒸发、干燥、在450 ℃条件下煅烧3 h得到Fe-VOx/SAPO-34催化剂。
采用浸渍法制备Fe-VOx/TiO2:取0.15 g的Fe-VOx充分溶解于50 mL去离子水中,加入1 g的纳米TiO2,充分搅拌后,旋转蒸发、干燥、在450 ℃条件下煅烧3 h得到Fe-VOx/TiO2催化剂。
钾中毒样品制备:将制备好的各催化剂加入质量分数为1%K2O溶液中,充分搅拌溶解后,旋转蒸发、干燥、在450 ℃条件下煅烧3 h得到K-Fe-VOx/SAPO-34和K-Fe-VOx/TiO2。
1.3 催化剂的表征
X射线衍射(XRD):采用日本Rigaku公司的TTRAX Ⅲ型X射线衍射仪来测定化剂晶相结构。测试电压40 kV,电流40 mA,在5°~80°范围内进行扫描。
X射线光电子能谱(XPS):采用日本岛津/Kratos AXIS SUPRA+型光电子能谱仪分析催化剂的表面元素价态,所得到的结果通过C 1s=284.6 eV进行校正。
程序升温还原(H2-TPR/NH3-TPD):采用美国麦克Autochem Ⅱ 2920仪器进行测试,将催化剂以10 ℃/min的速率升温至300 ℃预处理1 h后降温至50 ℃,将载气切换为10%H2/Ar (5%NH3),并在50 ℃下吹扫1 h,以10 ℃/min的升温速率升至900 ℃,并由检测器记录信号。
原位红外漫反射(in-situ DRIFTs):采用Nicolet iS50型傅里叶红外光谱仪进行测试,在100 mL/min N2气氛中,升温至400 ℃对催化剂进行预处理30 min,采集对应温度下的红外背景,通入1.0×10-4NH3(NO+O2)吹扫20 min,检测光谱。
1.4 催化剂活性测试
本实验中NH3-SCR活性测试均在VDRT-200SMT型微型多相催化反应器中进行。通入5.0×10-4NO、5.0×10-4NH3、5.0×10-2O2,并以氮气作为载气,气体总流量:260 mL/min。催化剂样品经过压片、过筛(40~60目),催化测试的体积空速为40 000 h-1。根据采集的数据样本,通过以下公式进行氮氧化物转化率(NO conversion)计算:
(1)
式中:[NO]in表示气体进口处的氮氧化物(NO和NO2)处于稳态时的浓度;[NO]out表示气体出口处氮氧化物处于稳态时的浓度。
2 结果与讨论
2.1 催化剂性能分析
图1为催化剂的NOx转化率随测试温度变化的曲线,Fe-VOx/SAPO-34和Fe-VOx/TiO2催化剂在240~420 ℃都具有90%以上的NOx转化率,在引入碱金属后,K-Fe-VOx/SAPO-34在260~420 ℃温度范围内仍能保持90%以上的催化活性,而K-Fe-VOx/TiO2催化剂在低温段出现失活现象。因此,将Fe-VOx负载在SAPO-34载体上表现出优异的抗碱金属能力。
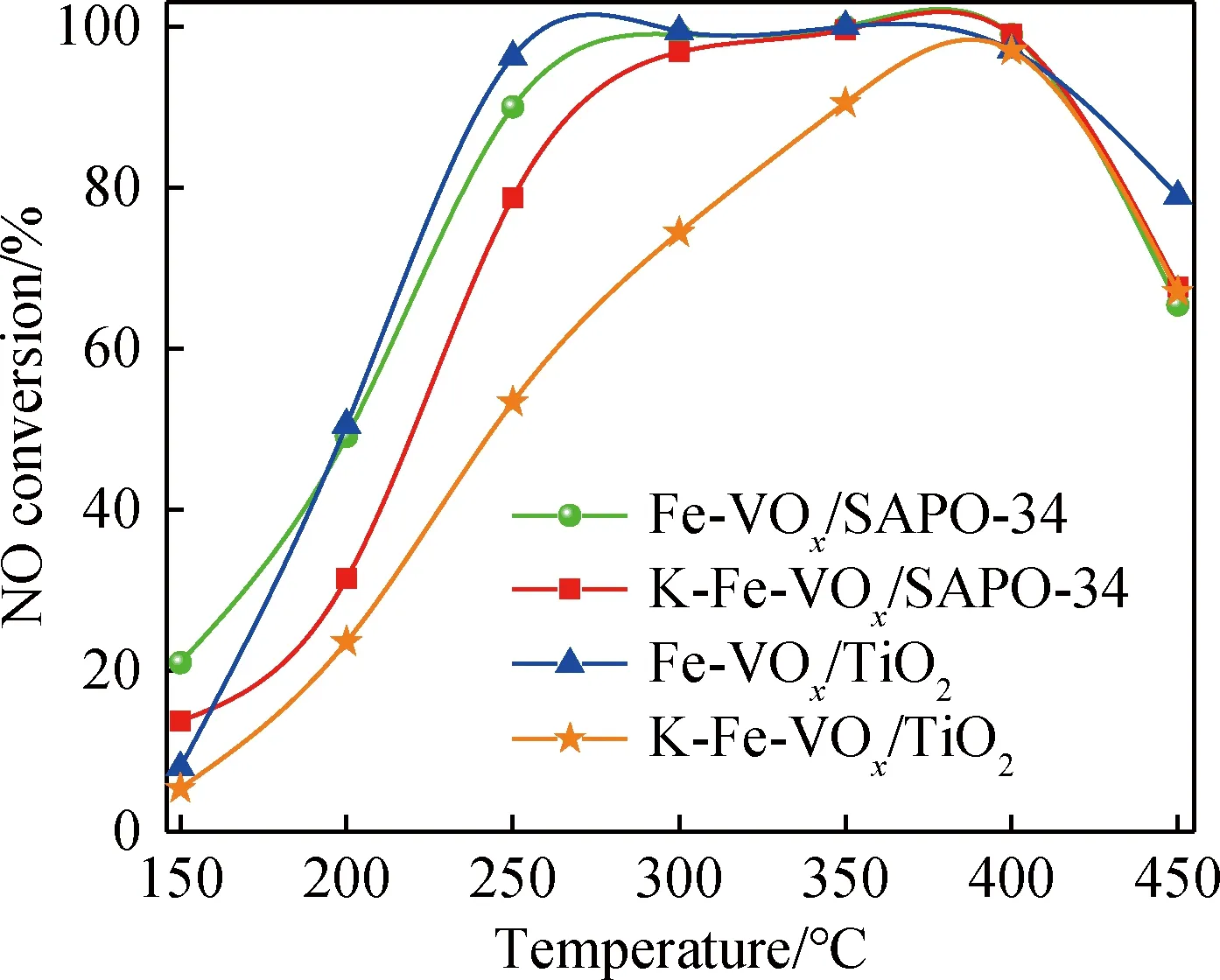
图1 催化剂在不同温度下的NO转化率Fig.1 NO conversion of catalyst at different temperatures
如图2所示,在240 ℃条件下对Fe-VOx/SAPO-34和Fe-VOx/TiO2催化剂的稳定性和抗水性能进行测试。测试过程中,Fe-VOx/TiO2催化剂的NOx转化率随时间的延长,催化活性逐渐降低,而Fe-VOx/SAPO-34催化剂对NOx转化率保持稳定,这主要是由于SAPO-34分子筛对表面活性物种的锚定作用,使得催化剂具有较强的稳定性。在反应过程中引入水后,结果显示两种催化剂在24 h反应时间内的NOx转化率没有明显变化,表明分子筛表面丰富的酸位能够抑制水汽和氨气的竞争吸附作用。以上结果表明,Fe-VOx/SAPO-34催化剂有良好的稳定性和耐水性,具有一定实际应用的潜力。

图2 催化剂的稳定性及抗水性能测试Fig.2 Stability and H2O resistance tests of catalysts
2.2 催化结构性质
催化剂的XRD测试结果如图3所示,制备样品中均未观测到明显的FeVO4特征衍射峰(PDF#71-1592),说明活性组分均匀分散于催化剂表面。同时Fe-VOx/SAPO-34催化剂衍射峰的峰形尖锐且半峰全宽较窄,表明样品具有良好的结晶性[17],浸渍负载制备过程及碱金属中毒过程未对SAPO-34的晶体结构产生明显影响。

图3 催化剂的XRD图谱Fig.3 XRD patterns of catalysts
2.3 催化表面物化性质
用XPS技术对催化剂表面元素比例进行分析,如图4、5、6和表1所示。其中,图4为催化剂的Fe 2p谱图,制备的催化剂均观察到Fe 2p3/2(710.3~710.9 eV和712.0~712.3 eV)和Fe 2p1/2(723.8~724.1 eV)的特征峰。在Fe-VOx系列催化过程中,Fe物种可参与表面的催化反应进程,且Fe3+相较于低价态的Fe具有更高的氧化还原性能[18]。通过表1可知,引入碱金属后,Fe-VOx/SAPO-34的Fe3+含量由63.35%减少到50.12%,而Fe-VOx/TiO2中Fe3+含量大量减少,由72.43%减少到39.21%。这是由于SAPO-34分子筛表面的酸位点有效降低碱金属对活性组分的侵害,使得催化剂表面氧化还原能力得以最大限度保留。

图4 催化剂的Fe 2p图谱Fig.4 Fe 2p spectra of catalysts
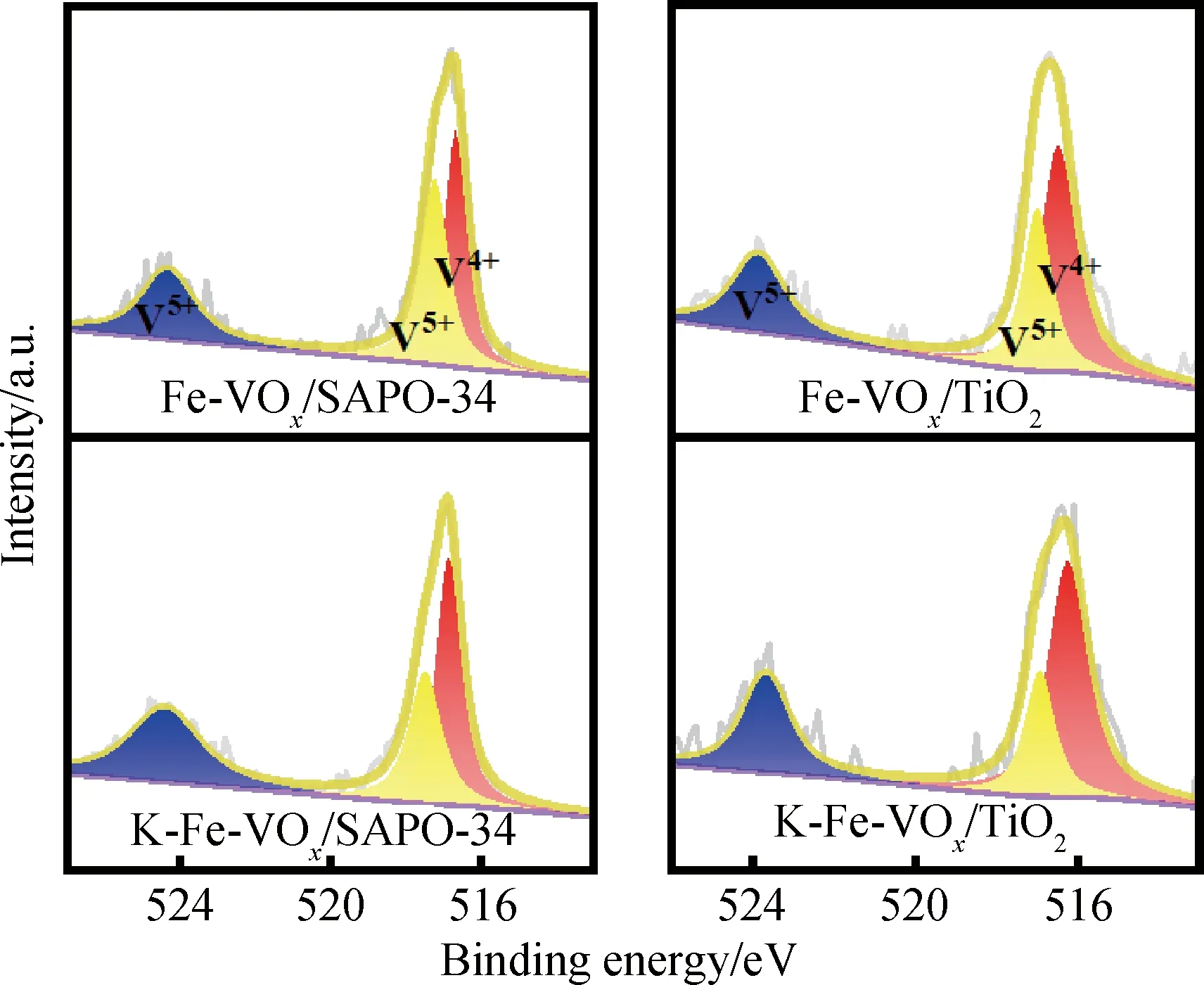
图5 催化剂的V 2p图谱Fig.5 V 2p spectra of catalysts

图6 催化剂的O 1s图谱Fig.6 O 1s spectra of catalysts

表1 催化剂表面元素相对含量Table 1 Relative content of each element on the surface
由图5中V 2p的XPS图谱可知,对于两种催化剂来说均可观察到V 2p3/2(516.2~517.6 eV)和V 2p1/2(523.7~524.5 eV)轨道,其中V 2p3/2轨道中516.2 eV归属于V4+2p3/2,而517.6 eV的峰归属于V5+2p3/2。由于碱金属的参与使Fe-VOx/SAPO-34催化剂属于V5+的峰减弱且向低结合能方向移动,这表明钾物种与钒物种发生键合作用,抑制了相应的活性位点,同样对于Fe-VOx/TiO2催化剂也是如此。
由图6中O 1s的XPS图谱可以看到,结合能在532.3~533.3 eV处的峰对应于化学吸附氧Oα,主要以O2、O-和O22-的形式存在,而在529.5~531.3 eV处的峰则对应于晶格氧Oβ,主要以O2-物种存在。化学吸附氧Oα具有更高的氧化性和移动性,有利于NO氧化为NO2及NH3的吸附和活化[19]。从表1中可知,催化剂中Oα含量按大小顺序排列为:Fe-VOx/SAPO-34>K-Fe-VOx/SAPO-34> Fe-VOx/TiO2>K-Fe-VOx/TiO2,这是因为SAPO-34分子筛表面的P、Al与Fe-VOx在颗粒界面处发生相互作用引入更多的氧空位[20-21],有助于增强NO的氧化并产生更多的NO2以促进催化剂“快速SCR”反应。
2.4 催化剂NH3吸附情况及氧化还原性能
图7为催化剂的NH3-TPD图谱,可以观察到Fe-VOx/SAPO-34催化剂的峰位于176 ℃和361 ℃,Fe-VOx/TiO2催化剂的峰位于250 ℃和353 ℃,其中Fe-VOx/SAPO-34脱附峰面积略微较大,表明催化剂表面存在更多的酸性位点,易于NH3的吸附。引入碱金属后,Fe-VOx/SAPO-34催化剂还原峰位置向前发生移动,且峰面积变小,这是由于SAPO-34表面的部分酸位点作为碱捕获位,与碱金属结合,使得表面吸附氨减弱。此外,从图中可知中毒前后Fe-VOx/SAPO-34催化剂的弱酸量相对没有明显变化,位于350 ℃左右的中强酸位点降低,而中毒后的Fe-VOx/TiO2催化剂酸量减少,这说明在弱酸位点与中强酸位点同时存在时,碱金属会优先损害中强酸位点,从而减少对弱酸位点的损害,使得中毒后Fe-VOx/SAPO-34催化剂具有良好的活性。

图7 催化剂的NH3-TPD图谱Fig.7 NH3-TPD patterns of catalysts
H2-TPR测试结果如图8所示,催化剂在410~478 ℃的峰归属于Fe3+→Fe2+与V5+→V4+的共还原峰,从图中可以看出,催化剂在低温段的峰形尖锐,说明活性组分在载体上均匀分散,这与XRD测试结果一致。同时,中毒前后的Fe-VOx/SAPO-34的还原峰面积小于Fe-VOx/TiO2和K-Fe-VOx/TiO2,这可能是Fe-VOx在煅烧过程中与SAPO-34分子筛载体发生一定固相反应,或与载体间发生强相互作用,导致部分物质难以还原[22-23]。此外,引入碱金属后,两种催化剂的还原峰向高温段发生偏移,导致催化剂还原能力下降,其中K-Fe-VOx/TiO2的变化尤为明显,还原峰由410 ℃偏移至478 ℃,这解释了K-Fe-VOx/TiO2中毒后失活的现象。
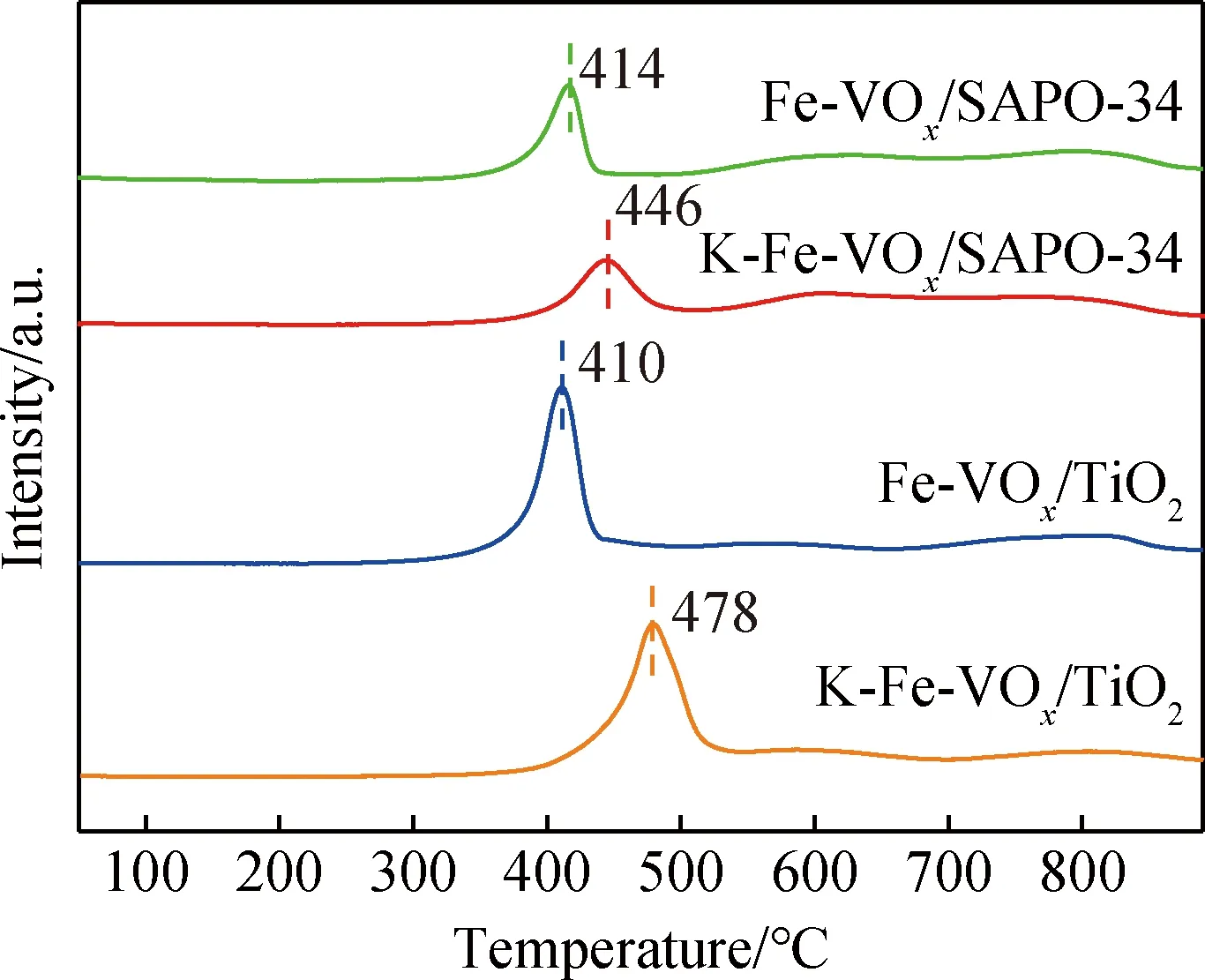
图8 催化剂的H2-TPR图谱Fig.8 H2-TPR patterns of catalysts
2.5 催化剂表面吸附原位红外研究
为了进一步研究K中毒后吸附在催化剂表面的NH3和NOx物种对催化剂表面反应过程的影响,对中毒前后Fe-VOx/SAPO-34的in-situ DRIFTs光谱进行分析,如图9所示。对于Fe-VOx/SAPO-34脱硝催化剂,饱和吸附NH3后,在1 214 cm-1处出现吸附在Lewis酸位点配位NH3中的N—H的对称和不对称弯曲振动引起的特征峰,1 447 cm-1和1 728 cm-1处的峰属于Brønsted酸位点上NH4+物种的伸缩震动[24],其峰强度较强,这得益于SAPO-34分子筛提供了较多的酸性位点。引入碱金属后,催化剂整体吸脱附没有较大影响,仅是属于Brønsted酸位点上NH4+物种的峰信号减弱,进而可以说明催化剂表面部分Brønsted酸位点可以优先形成牺牲位与碱金属结合,而剩余的酸位能够维持SCR反应的进行。

图9 催化剂的NH3吸脱附原位红外光谱图及相应的mapping图。(a)(b)Fe-VOx/SAPO-34; (c)(d)K-Fe-VOx/SAPO-34Fig.9 In situ DRIFTS of NH3 desorption catalysts and corresponding mapping results.(a)(b) Fe-VOx/SAPO-34; (c)(d) K-Fe-VOx/SAPO-34
图10所示的NO+O2吸脱附实验结果表明,Fe-VOx/SAPO-34脱硝催化剂吸附NO+O2后,在1 405 cm-1、1 446 cm-1和1 619 cm-1处出现特征峰。其中位于1 405 cm-1和1 446 cm-1的峰属于双齿硝酸盐物种,而处于1 619 cm-1的峰为吸附态NO2物种不对称弯曲振动。钾中毒后,双齿硝酸盐物种消失,吸附态NO2物种的峰强度发生减弱。其中,在1 213 cm-1处出现了桥联硝酸盐物种,而桥连硝酸盐物种活泼性弱于双齿硝酸盐物种[25],从而导致脱硝活性降低。综上所述,引入碱金属会对吸附态NO2物种与双齿硝酸盐物种的生成产生抑制作用,并产生部分惰性物种,从而对催化剂的SCR性能产生一定影响。
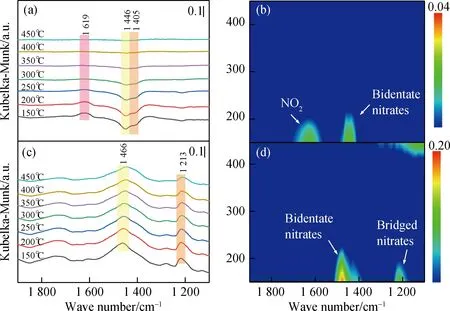
图10 催化剂的NO+O2吸脱附原位红外光谱图及相应的mapping图。(a)(b)Fe-VOx/SAPO-34; (c)(d)K-Fe-VOx/SAPO-34Fig.10 In situ DRIFTS of NO+O2 desorption catalysts and corresponding mapping results.(a)(b) Fe-VOx/SAPO-34; (c)(d) K-Fe-VOx/SAPO-34
2.6 催化剂表面原位暂态红外反应


图11 催化剂在180 ℃下,先吸附NO+O2再吸附NH3的原位暂态反应及相应的mapping图(a)(b)Fe-VOx/SAPO-34; (c)(d)K-Fe-VOx/SAPO-34Fig.11 In situ DRIFTS of the transient reactions at 180 ℃ for catalyst between NH3 and pre-adsorbed NO+O2 (a)(b) Fe-VOx/SAPO-34; (c)(d) K-Fe-VOx/SAPO-34

图12 催化剂在180 ℃下,先吸附NH3再吸附NO+O2的原位暂态反应及相应的mapping图(a)(b)Fe-VOx/SAPO-34; (c)(d)K-Fe-VOx/SAPO-34Fig.12 In situ DRIFTS of the transient reactions at 180 ℃ for catalyst between NO+O2 and pre-adsorbed NH3(a)(b) Fe-VOx/SAPO-34; (c)(d) K-Fe-VOx/SAPO-34
3 结 论
采用浸渍法制备Fe-VOx/SAPO-34催化剂,克服了Fe-VOx负载TiO2载体催化剂脱硝性能易受碱金属影响等问题。在引入碱金属后,Fe-VOx/SAPO-34在260~420 ℃内仍保持90%以上的NOx转化率。对催化剂进行一系列表征测试,发现碱中毒过程未对SAPO-34分子筛结构产生破坏,其表面的P、Al与Fe-VOx发生相互作用能够引入更多的氧空位,提高催化剂的氧化还原性能。此外,碱金属能够被SAPO-34分子筛上丰富的酸位点捕获,减弱钾对活性组分的侵害,保证催化反应的高效进行。
