遗传性球形红细胞增多症合并Gilbert综合征1例报告
2022-04-14张玉姣王秀红马安林
张玉姣, 郑 英,2, 王秀红, 蔡 颖, 马安林
1 中日友好医院 a.感染疾病科, b.病理科, c.检验科, 北京 100029;2 解放军联勤保障部队第九四〇医院 消化内科, 兰州 730050
1 病例资料
患者男性,26岁,因“反复全身皮肤及巩膜黄染26年”于2021年6月24日收住中日友好医院。患者在出生5个月时出现全身皮肤、巩膜黄染,尿色加深,未予重视。12岁时,再次因“皮肤及巩膜黄染”就诊于解放军总医院,腹部超声提示:慢性胆囊炎、胆囊结石(充满型)、脾肿大,未明确诊断。后就诊于北京儿童医院,查肝功能:DBil 38 μmol/L,IBil 206.3 μmol/L,ALT、AST、GGT、ALP均正常,血常规:WBC 11.7×109/L, RBC 2.53×1012/L, Hb 70 g/L,红细胞平均体积 74 fL, PLT 296×109/L,网织红细胞 11.23%,外周血涂片:可见球形红细胞;红细胞渗透脆性试验:开始溶血>0.66%(0.42%~0.46%),完全溶血0.40%(0.32%~0.34%),酸化甘油试验时间50 s(>290 s),红细胞葡萄糖-6-磷酸脱氢酶活性正常,血红蛋白F碱变性试验正常:0.85(不支持地中海贫血),直接和间接抗人球蛋白试验阴性,诊断为遗传性球形红细胞增多症,行脾切除术,保胆取石术,术后患者仍有反复全身及巩膜轻度黄染。本次住院前入职体检肝功能提示:TBil 55.24 μmol/L,DBil 13.28 μmol/L、IBil 41.96 μmol/L,腹部超声提示副脾4 cm,为进一步诊治门诊以“黄疸待查”收住中日友好医院感染疾病科。患者无皮肤瘙痒,生长发育及日常生活均正常,直系亲属中无类似情况。入院查体:生命体征平稳,全身皮肤、巩膜轻度黄染,无肝掌及蜘蛛痣,心肺未查及明显异常,腹部平软,左上腹可见长约8 cm横形陈旧手术瘢痕,肝脾未触及,墨菲征阴性,麦氏点压痛阴性,肠鸣音正常,移动性浊音阴性,双下肢无凹陷性水肿。血常规:WBC 9.66×109/L、RBC 4.8×1012/L、Hb 149 g/L、红细胞平均体积 84.4 fL,红细胞体积分布宽度47.7 fL、PLT 342×109/L,网织红细胞3.56 %(0.5%~1.5%);外周血涂片:红细胞轻度大小不等,球形红细胞约占28%(图1);肝功能:ALT 13 U/L、AST 15 U/L、TBil 71.37 μmol/L、DBil 20.71 μmol/L、ALP 41 U/L、GGT 17 U/L;LDH 191(100~250) U/L,尿含铁血黄素试验、尿常规、粪便常规、AFP、凝血、免疫球蛋白、HBsAg、抗-HCV、抗-HAV-IgM、抗-HEV-IgM、抗CMV-IgM、抗EBV-IgM、自身免疫性肝病抗体谱、抗核抗体谱均正常。患者黄疸特点是以IBil升高为主,且目前Hb正常,故行UGT1A1基因检测(图2),结果示:(1)患者UGT1A1基因启动序列插入变异,使(TA)6TAA变异到(TA)7TAA,即由野生型UGT1A1*1变异到UGTA1*28;(2)位于外显子(exon)1的错义变异,c.211G>A,即UGT1A1*6杂合。启动子UGT1A1*28杂合突变与外显子UGT1A1*6共同杂合突变导致葡萄糖醛酸转移酶的活性降低,确诊为Gilbert综合征。同时行肝穿刺,病理提示(图3):中央静脉周围少部肝细胞内见脂褐素沉积,符合Gilbert综合征。综合患者的所有病例资料,诊断为遗传性球形红细胞增多症合并Gilbert综合征。
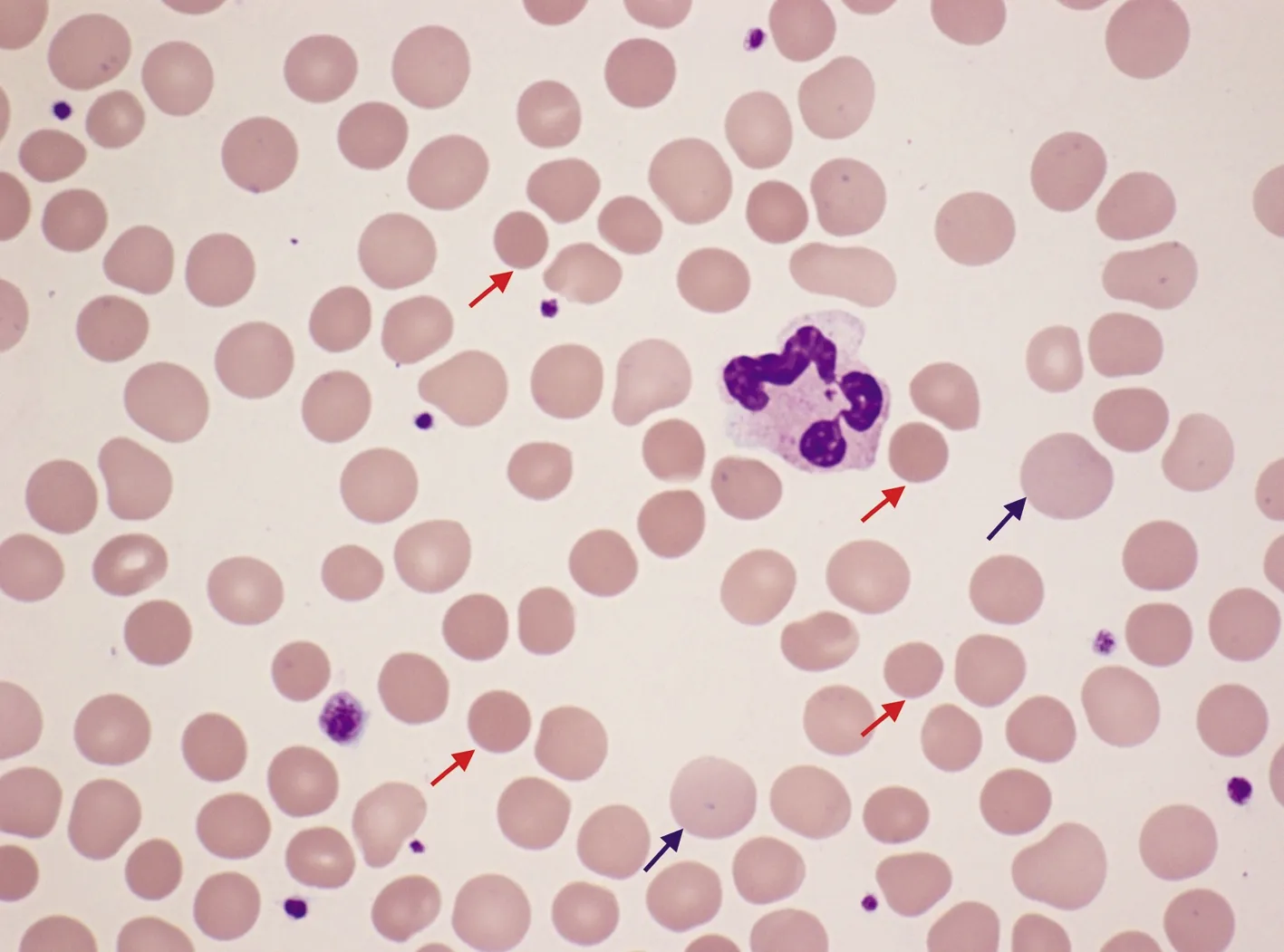
注:红色箭头示球形红细胞,表现为密集的球状红细胞,中央淡染区丢 失;蓝色箭头示多染色性网织红细胞。
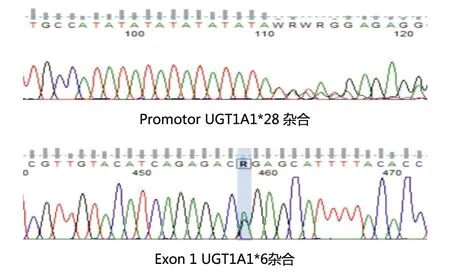
图2 基因检测结果Figure 2 Genetic testing results

注:中央静脉周围部分肝细胞内见脂褐素沉积。图3 肝脏病理结果(HE染色,×400)Figure 3 Liver pathological results (hematoxylin-eosin staining,×400)
2 讨论
遗传性球形红细胞增多症多为常染色体显性遗传,少数为常染色体隐性遗传,人群发病率约为1/2000,遗传性球形红细胞增多症是位于第6号或第8号染体基因突变,由于红细胞的膜缺陷使其形态呈球形,在通过脾脏的时候被大量破坏,从而导致IBil升高为主的溶血性黄疸,典型的临床表现为贫血、黄疸、脾大,部分患者伴有胆石症[1]。Gilbert综合征通常认为是常染色体隐性遗传,也有研究认为它是常染色体显性遗传,具有不完全外显率,人群发病率为2%~20%,Gilbert综合征是位于2号染色体的尿苷二磷酸葡萄糖醛酸转移酶(uridine diphosphate glucuronosyl transferase, UGT)1A1基因突变,使UGT活性下降,因而肝细胞对IBil的处理能力下降,引起以IBil升高为主的先天性非溶血性黄疸,主要临床表现为慢性、间歇性高间接胆红素血症,无溶血及肝脏疾病表现[2-3]。遗传性球形红细胞增多症与Gilbert综合征是由不同基因突变引起的遗传性疾病,虽同时罹患的概率极低,但国内外均有报道,仍需引起临床重视[4]。
高间接胆红素血症常见于溶血性黄疸及UGT1A1基因突变[5]。UGT1A1基因突变所致的疾病,主要是Gilbert综合征和Crigler-Najjar综合征(Crigler-Najjar syndrome,CNS)[6-7]。按UGT1A1活性划分,Gilbert综合征患者肝脏中的UGT1A1活性约为正常人的30%[8],CNS Ⅱ型UGT1A1活性约为正常人的10%,CNS Ⅰ型UGT1A1活性大多缺失或低于正常人的1%[6]。溶血性黄疸的TBil水平常不高于85.5 μmol/L[9],Gilbert综合征患者的TBil水平为17.1~102. 6 μmol/L,CNS Ⅱ型患者TBil水平为102. 6~342 μmol/L,CNS Ⅰ型患者TBil水平为342~769. 5 μmol/L[10]。该患者12岁时在儿童医院查DBil 38 μmol/L,IBil 206.3 μmol/L,远高于遗传性球形红细胞增多症所致的溶血性黄疸的胆红素水平。虽然患者明确诊断为遗传性球形红细胞增多症并行脾切除术,但是随诊发现患者黄疸始终存在,本次住院查TBil 71.37 μmol/L、DBil 20.71 μmol/L。患者网织红细胞比例增多,提示患者仍有溶血;但是患者脾脏已经切除,血常规中RBC、Hb基本正常,LDH在正常范围内,说明溶血程度并不严重,此时的溶血程度与目前TBil、IBil升高的比例也不相符,单纯用遗传性球形红细胞增多症所致的溶血性黄疸不能完全解释。高间接胆红素血症还常见于UGT1A1基因突变。对该患者的UGT1A1基因进行筛查,发现患者存在UGT1A1*28与UGT1A1*6共同杂合突变,这也是Gilbert综合征在我国汉族中较为广泛的突变基因[11]。综上,考虑患者14年前出现的极高程度的IBil是由肝前性IBil生成过多(红细胞破坏增加)及肝性IBil转化减少(UGT1A1基因突变)共同引起。
从该病例的诊断过程中总结,高间接胆红素血症要结合患者的临床表现,综合筛查患者是否存在溶血及UGT1A1基因异常。溶血性黄疸要考虑到遗传性溶血性贫血、获得性溶血性贫血,UGT1A1基因异常要考虑到Gilbert综合征、CNS。当胆红素升高水平与所考虑疾病不相符时,要注意是否存在多种疾病的重合。对于遗传性球形红细胞增多症的患者,脾切除是有效的治疗方法,术后球形红细胞虽依然存在,但红细胞寿命延长,可使黄疸减轻、Hb上升[1]; Gilbert综合征目前认为是良性病变,暂无需特殊治疗[2]。由于遗传性球形红细胞增多症和Gilbert综合征都是遗传性疾病,因此给予患者及时、正确的诊断,可给他们未来的优生优育提供指导。
伦理学声明:本例报告已获得患者知情同意。
利益冲突声明:所有作者均声明不存在利益冲突。
作者贡献声明:张玉姣、郑英参与病例资料分析并起草文章;王秀红、蔡颖、马安林参与修改文章关键内容。
