双盐镁电池CoS2正极材料的电化学性能研究
2022-04-12李文博黄民松李月明李驰麟
李文博, 黄民松, 李月明, 李驰麟,4
双盐镁电池CoS2正极材料的电化学性能研究
李文博1,2, 黄民松2,3, 李月明1, 李驰麟2,3,4
(1. 景德镇陶瓷大学 材料科学与工程学院, 景德镇 333403; 2. 中国科学院 上海硅酸盐研究所, 高性能陶瓷和超微结构国家重点实验室, 上海 201899; 3. 中国科学院大学 材料与光电研究中心, 北京 100049; 4. 中国科学院 上海硅酸盐研究所, 中国科学院能量转换材料重点实验室, 上海 201899)
镁金属电池因为镁金属负极的高体积比容量(3833 mAh/cm3)和高安全性而日益受到关注。然而, Mg2+引起的极化效应抑制了Mg2+在固相中的扩散, 限制了镁金属电池的比容量。锂镁双盐电解液利用Li+代替Mg2+驱动正极反应, 能够绕开Mg2+在固相中扩散缓慢的问题。本工作研究了过渡金属硫化物CoS2在不同锂镁混合电解液中的电化学性能, 并分析了锂盐浓度和充放电电压区间对其转换反应和循环稳定性的影响。添加锂盐的策略提高了CoS2基镁金属电池的转换反应动力学, 当充电电位提高至2.75 V时, Mg-CoS2电池在LiCl-APC电解液中的循环稳定性得到显著提高, 在循环150次后, 其比容量仍能维持在275 mAh/g, 远高于在2.0 V截止电压条件下的33 mAh/g。电池容量衰减与CoS2正极在2.0 V充电电位下Co3S4的不可逆生成有关, 其长期循环中伴随的Co和S元素溶解加剧了容量的不可逆损失。本工作为过渡金属硫族化合物在转换反应型镁电池中的应用提供了一种激活策略。
镁金属电池; CoS2; 锂镁双盐体系; 正极材料; 转换反应
由于石墨负极理论比容量较低(372 mAh/g), 锂离子电池难以满足储能系统对能量密度日益增长的需求[1]。而锂金属电池因锂金属负极具有极高的理论比容量(3860 mAh/g)和最低的电化学电位(–3.04 V (. SHE)), 被认为是下一代高比能储能电池的有力竞争者[2-4], 然而, 不可控的锂枝晶生长问题极大限制了锂金属电池的发展。锂枝晶会消耗大量的活性金属锂和电解液, 导致电池性能迅速衰减, 而且会刺穿隔膜, 引发热失控等安全隐患[3-4]。
作为后锂离子电池时代的候选之一, 镁金属电池由于镁资源丰富、理论比容量高和安全性好等优点而受到越来越多的关注[5-7]。金属镁储量丰富, 价格不到金属锂的1/30, 发展二次镁金属电池可以有效降低电池成本[5]。与金属锂(2066 mAh/cm3)相比, 镁金属的体积比容量高达3833 mAh/cm3[6]; 而且其电化学电位仅为–2.37 V (. SHE), 与适当的正极材料相匹配时能量密度十分可观。更重要的是, 相比于金属锂, 金属镁在电化学沉积/剥离过程中不会形成枝晶, 更为安全可靠[7]。此外, 由于金属镁本身化学性质不活泼, 能在空气中稳定存在, 因此镁用作电池负极时电池的可操作性增强[8]。基于以上原因, 镁金属电池被认为是一种有望用于电动汽车和大型储能装置的新型绿色二次电池。
虽然镁金属在成本和容量上有潜在的优势, 但缺乏合适的电解液和正极材料, 这极大制约了镁金属电池的发展[9]。金属镁与大多数电解液不相容, 电解液会在镁金属表面被还原, 导致致密钝化层的生成, 阻碍Mg2+的传输[10-11]。此外, 虽然Mg2+(0.086 nm)和Li+(0.09 nm)的离子半径相近, 但由于Mg2+带有两个电荷, 电荷密度远大于Li+, 所产生的极化效应和溶剂化作用也远大于Li+, 导致Mg2+在固相中扩散缓慢, 因此很难找到适合的能实现可逆嵌入/脱出Mg2+的正极材料[12-13]。为了克服Mg2+扩散缓慢的问题, 研究者提出了锂镁双盐混合电解液的概念, 其特征是电解液中同时含有锂盐和镁盐, 负极发生金属镁的可逆沉积/析出过程, 正极的电化学反应则由电解液中的Li+驱动, 避免了Mg2+在正极材料中扩散缓慢的问题[14-16]。正极材料的选择范围得到大大拓展, 例如基于插层反应的Li4Ti5O12[16]、Mo6S8[17]、LiFePO4[18]、TiS2[19]、TiO2[20]和MoO2[21]等均可作为双盐体系的正极。然而基于插层反应正极材料的可逆容量有限(<150 mAh/g), 其能量密度通常低于300 Wh/kg[22]。采用基于转化反应型的正极材料能明显提高电池的可逆容量和能量密度, 例如金属硫化物[15,23-24], 单质S[6,25]和单质Se[26]等。如本课题组之前报道的基于LiCl-APC电解液的FeS2正极, 其能量密度达到400 Wh/kg[15]。因此, 将金属硫化物作为锂镁混合体系的正极材料, 利用Mg负极无枝晶的特点, 有望获得安全、稳定且高能量密度的镁金属电池。与其他金属硫化物相比, CoS2具有高电导率和良好的热稳定性, 已被应用于锂离子电池、半导体和催化等领域[27-29]。基于此, 本工作进一步研究了CoS2在锂镁混合电解液中的电化学性能, 对比分析了不同双盐混合电解液以及充放电电压区间对CoS2电化学性能的影响, 探讨了CoS2正极性能衰减的原因, 为金属硫化物在锂镁混合体系中的发展提供了数据支撑。
1 实验方法
1.1 Co3S4的水热法制备
将0.75 mmol CoCl2∙6H2O和7.5 mmol硫脲加入到40 mL去离子水中, 搅拌至完全溶解后转移到50 mL的水热釜中, 升温至180 ℃保温20 h, 待冷却至室温后, 离心收集固体并60 ℃真空干燥。
1.2 电解液配置
采用三种不同类型的双盐混合电解液对CoS2的电化学性能进行测试。(1)0.25 mol/L AlCl3(Sigma Aldrich)和0.5 mol/L苯基氯化镁(Aladdin)的四氢呋喃(THF, Alfa Aesar)溶液, 称为APC电解液; 0.5、1.0和1.5 mol/L LiCl (Sigma Aldrich), 0.25 mol/L AlCl3(Sigma Aldrich)和0.5 mol/L苯基氯化镁(Aladdin)的四氢呋喃(THF, Alfa Aesar)溶液, 称为LiCl-APC混合双盐电解液; (2) 1.0 mol/L LiBH4(J&K)和0.1 mol/L Mg(BH4)2(Sigma Aldrich)的二甘醇二甲醚(DGM, Sigma Aldrich)溶液, 称为LiBH4-Mg(BH4)2/DGM混合电解液; (3)1.0 mol/L双三氟甲磺酰亚胺锂(LiTFSI, Sigma Aldrich), 0.1 mol/L双(六甲基二硅叠氮)镁(Mg(HMDS)2, Sigma Aldrich), 0.2 mol/L AlCl3和0.3 mol/L MgCl2(Alfa Aesar)的DGM溶液, 称为LiTFSI-MHCC/DGM混合电解液。
1.3 电化学性能测试
将CoS2(Alfa Aesar)和合成的Co3S4粉末分别与Super P导电炭黑以及聚偏氟乙烯(PVDF)按质量比7 : 2 : 1 混合, 研磨均匀后加入适量的-甲基吡咯烷酮(NMP)溶剂继续研磨。将研磨均匀的浆料涂覆在10 mm的碳纸上, 然后在60 ℃真空干燥12 h得到CoS2和Co3S4正极电极片, 活性物质的载量约为1.3 mg/cm2。将其与金属镁负极、glass fiber隔膜和不同类型的双盐混合电解液组装成CR2032扣式电池。室温下, 在LAND电池测试系统中进行电化学测试。
1.4 表征
使用X射线衍射仪(XRD, D8, Bruker)分析CoS2和合成的Co3S4的晶体结构。利用透射电子显微镜(TEM, JEOL JEM-2100F)观察CoS2正极在充放电后的形貌和微观结构。用扫描电子显微镜(SEM, SU8200)表征镁金属负极在循环后的形貌和元素组成。使用X射线光电子能谱(XPS, ESCAlab-250)分析初始和循环后CoS2样品的表面组成和价态。
2 结果与讨论
2.1 CoS2的电化学性能
图S1(a)为商业CoS2粉末的XRD图谱, 其峰形尖锐, 结晶度良好, 与立方相CoS2(PDF 89-1492)的标准衍射数据一致。图S1(b)为CoS2正极循环前的SEM照片, 其中CoS2活性物质呈现球形, 与导电炭分布均匀, 粘结剂PVDF很好地连接了CoS2颗粒与导电炭颗粒, 颗粒紧密堆积在一起, 形成了良好的导电网络。为了确定混合电解液中锂盐的正面作用, 首先测试CoS2正极在纯APC电解液中的性能。如图1(a)所示, 当不添加锂盐时, CoS2正极的首圈比容量仅有28 mAh/g, 随后的可逆容量几乎可忽略不计。从图S2的充放电曲线可以看出, CoS2正极在纯镁电解液中的电压极化很大, 证实高电荷密度的Mg2+会降低其在固相中的扩散速率, 导致正极反应动力学不佳[12]。图1(a,b)是CoS2正极在不同锂镁混合电解液中的长循环性能和充放电曲线对比。加入锂盐能提高镁金属电池的反应动力学, 在0.1(1=1.13 mA/cm2)倍率下, 基于LiCl-APC、LiBH4-Mg(BH4)2以及LiTFSI-MHCC电解液的Mg-CoS2电池的初始放电比容量分别为809、855和604 mAh/g, 远高于纯APC电解液中的初始比容量28 mAh/g。然而, 锂镁混合电解液虽然能激活Mg-CoS2电池的初始动力学, 但其在不同混合电解液中均表现出前期容量衰减。循环100次后, 它们的放电比容量分别为44、82和104 mAh/g, 容量衰减率分别达到94.5%、90.4%和82.8%。而CoS2正极在LiCl-APC电解液中的容量衰减更迅速, 为分析放电比容量衰减的原因, 本研究以LiCl-APC电解液体系为研究对象, 分析锂盐浓度和充放电压区间对CoS2正极性能的影响。
Mg-CoS2电池在含有不同浓度锂盐的LiCl-APC电解液中表现出相似的衰退性。如图1(c,d)所示, 在0.5 mol/L锂盐的LiCl-APC电解液中, 其初始放电比容量为719 mAh/g, 循环100和150次后, 其放电比容量衰减到31和27 mAh/g。与含低浓度锂盐的混合电解液的性能类似, 在1.0和1.5 mol/L锂盐的LiCl-APC电解液中, CoS2正极的首圈放电比容量虽然能提高至809和784 mAh/g, 但电池的循环稳定性仍然较差, 循环150次后, 其放电比容量分别仅剩33和32 mAh/g。上述结果证明锂盐能提高镁金属电池的反应动力学, 但Mg-CoS2电池在常用的充放电电压区间(0.1~2.0 V)仍表现出较差的循环稳定性, 且与混合电解液的种类及锂盐的浓度无关。
为进一步探究Mg-CoS2电池性能衰减的原因, 基于1.0 mol/L LiCl-APC电解液, 通过改变充电电压分析充放电压区间对CoS2正极性能的影响。在上述0.1~2.0 V电压区间基础上, 进一步将充电电压拓宽至2.75 V, 结果如图2(a)所示。一般来说, 当充电电压超过2.0 V时, 电解液中的Cl–会腐蚀不锈钢集流体[30]。为避免充电电压过高所带来的副反应, 可以用钼片替换不锈钢作为电池的集流体, 防止电解液对电池的腐蚀[31]。当充电电压提高至2.75 V后, Mg-CoS2电池的循环稳定性得到极大提升, 循环150次后, 其放电比容量能维持在275 mAh/g, 远高于充电至2.0 V的放电比容量(33 mAh/g)。这与Li-CoS2锂金属电池的性能相当(图S3), 例如, Li-CoS2电池循环20圈后的放电比容量为~440 mAh/g, 而Mg-CoS2电池经20圈循环后的放电比容量为~450 mAh/g。高充电电位显著提高了CoS2在锂镁混合体系中的电化学循环性能。

图1 CoS2在不同混合电解液中基于0.1C和0.1~2.0 V充放电区间的(a)长循环性能及(b)相应的充放电曲线; CoS2在含有不同浓度锂盐的LiCl-APC电解液中的(c)循环性能及(d)相应的充放电曲线

图2 (a)在0.1C倍率下, 基于LiCl-APC电解液的Mg-CoS2电池在不同充放电区间的长循环性能对比; (b)基于LiCl-APC电解液的Mg-CoS2电池在不同充放电区间的倍率性能
为了测试大电流密度下的充放电特性, 研究对CoS2正极进行了倍率测试, 结果如图2(b)所示。通过在0.1、0.2、0.5、1和0.1倍率下各充放电10次, 能看出在0.1倍率, 不同电压区间下的首圈放电比容量接近(>720 mAh/g), 然后在窄电压区间的容量迅速衰减至~300 mAh/g。然而在0.2~1倍率下, 基于2.75 V充电电压的Mg-CoS2电池具有更高的放电比容量; 尤其在1大倍率下, 当充电电压提高至2.75 V后, 其放电比容量能维持在210 mAh/g, 而基于2.0 V充电电压的放电比容量仅有8 mAh/g。即便倍率从1回到0.1, 高充电电压电池的放电比容量仍能维持在~300 mAh/g, 而低压充放电的电池的比容量会衰减到~100 mAh/g。上述结果表明, 提高充电电压能显著提升CoS2正极在锂镁混合电解液中的循环稳定性和倍率性能, 充放电电压区间是影响CoS2正极循环稳定性的主要因素。
2.2 反应机理分析
为研究CoS2正极在混合电解液中的反应机理和性能衰减原因, 利用XPS和TEM对初始样品、首次放电和首次充电至2.0及2.75 V的样品进行元素价态和组成结构分析。如图3(a)的XPS能谱所示, 原始CoS2的Co2p能谱含有两个分别位于779.0和794.0 eV的Co2p3/2和Co2p1/2核心峰[32]。当放电至0.1 V后, Co2p3/2峰的结合能降至778.5 eV, 对应于单质Co[33]。值得注意的是, 当首次充电至2.0 V后, Co2p3/2峰的结合能提高至780.3 eV, 说明充电产物发生了变化。而当充电至2.75 V后, Co2p3/2峰的结合能与原始CoS2的结合能一致。从图3(b)的S2p能谱进一步看出, 放电至0.1 V后, 不仅生成了单质Co, 而且由于电解液中的Li+驱动的正极反应, 放电产物还含有Li2S[15]。而充电至2.0 V的样品的S2p能谱中不仅含有少量CoS2, 而且含有大量Co3S4, 其Co2p3/2和Co2p1/2峰分别位于161.4和162.6 eV; 此外, Co3S4峰更强, 表明其含量更大[34]。而当充电电压提升至2.75 V时, 其氧化产物的S2p能谱则由位于162.2和163.4 eV的Co2p3/2和Co2p1/2峰以及位于163.15和164.35 eV的Co2p3/2和Co2p1/2峰组成, 与原始CoS2的S2p能谱一致, 其中峰强的变化可归因于循环后电极的表面覆盖物。CoS2正极在较低充电电位下的不可逆行为是电池容量衰减的主要原因, 提高充电电压有利于改善正极材料的结构可逆性。
图4是CoS2正极在首次放电和充电至2.0及2.75 V后的TEM照片。如图4(a, b) 所示, 在深放电状态下(0.1 V), 放电产物不仅包含单质Co, 而且存在Li2S, 证实正极反应是由电解液中的Li+驱动的, 因而锂镁混合电解液能够提高正极的反应动力学。充电至2.0 V后, 其氧化产物主要由Co3S4而非CoS2构成, 这表明较低的充电电位不足以使首次充电产物回到初始状态。而当提高充电电位至2.75 V时, 充电产物进一步氧化生成CoS2。转换反应的可逆性保证了CoS2正极在锂镁混合电解液中的循环稳定性, 这与XPS的分析结果一致。同时, 循环后的电极活性颗粒的形貌维持良好, 转换反应并没有引起转换产物晶域离散, 但转换反应造成的体积变化使大颗粒内部出现连通的多孔, 这有利于电解液渗透, 而不影响晶域间的电接触。
CoS2正极在窄电位区间下的首次放电比容量虽然可观, 但其容量衰减抵消了锂镁混合电解液对CoS2正极的激活效果。XPS和TEM结果证实, 在低截止电位的充放电过程中, CoS2正极的不可逆相变是容量衰减的主要原因。通过镁金属负极表面的EDS能谱(图5)进一步看出, 循环50圈后镁金属表面不仅含有Mg、O、Cl, 还有S元素, 进一步表明正极侧CoS2在低充电电位下的不可逆转换伴随有多硫化物溶解。值得注意的是, 镁金属表面还含有Co元素, 这表明正极侧的CoS2中Co元素在充放电过程中也会逐渐溶解到电解液中, 从而迁移至负极侧, 造成正极侧活性物质的不可逆损失, 导致容量持续性衰减。从长期循环后的SEM照片可以看出, 镁负极表面确实不易形成枝晶, 电镀的镁金属仍然表现出大块状的形貌且接触紧密。
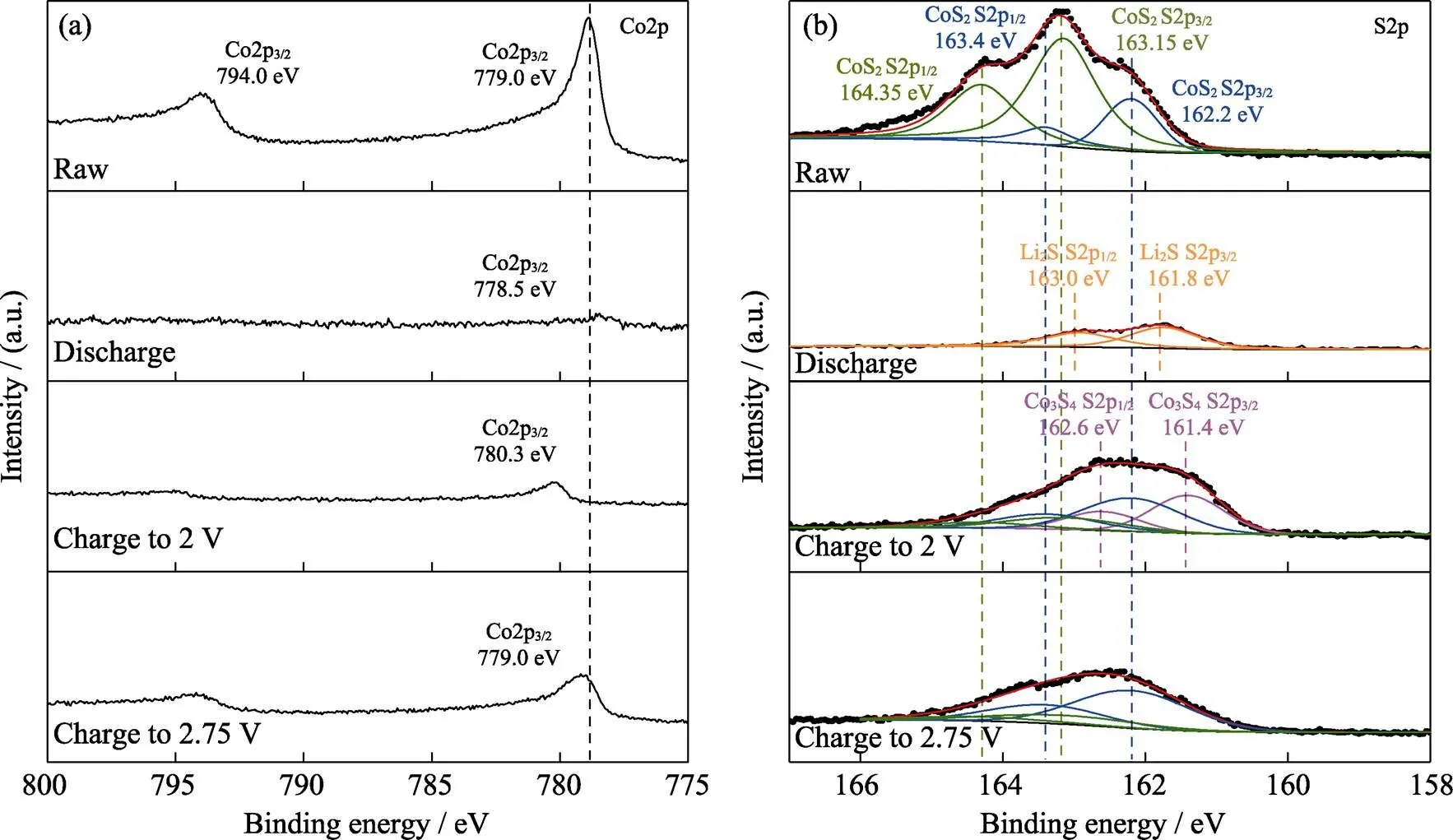
图3 CoS2原始电极、首次放电电极和首次充电至2.0和2.75 V电极的XPS(a)Co2p和(b)S2p能谱

图4 (a, b)首次放电后, 首次充电至(c, d)2.0和(e, f)2.75 V后CoS2电极的(a, c, e)TEM和(b, d, f)高分辨TEM照片

图5 (a)Mg-CoS2电池在LiCl-APC电解液中50次循环后的镁负极表面的SEM照片, 以及(b~f)对应的元素分布图
基于上述分析可知, 如图6所示, 在放电过程中, 正极反应由电解液中的Li+主导, CoS2被还原成单质Co和Li2S, 而负极侧则发生镁金属的剥离。在低截止电位条件下的充电过程中, 正极侧的单质Co和Li2S发生反应生成Co3S4而非初始的CoS2, 并伴随少量Co和S元素的溶解, 而负极侧则发生镁金属的沉积。CoS2正极在低电位区间首次充放电的电化学反应机理可概括为:
首次放电:
CoS2+4Li++4e–→Co0+2Li2S (1)
Mg–2e–→Mg2+(2)
首次充电至2.0 V:
3Co0+4Li2S→Co3S4+8Li++8e–(3)
Mg2++2e–→Mg (4)
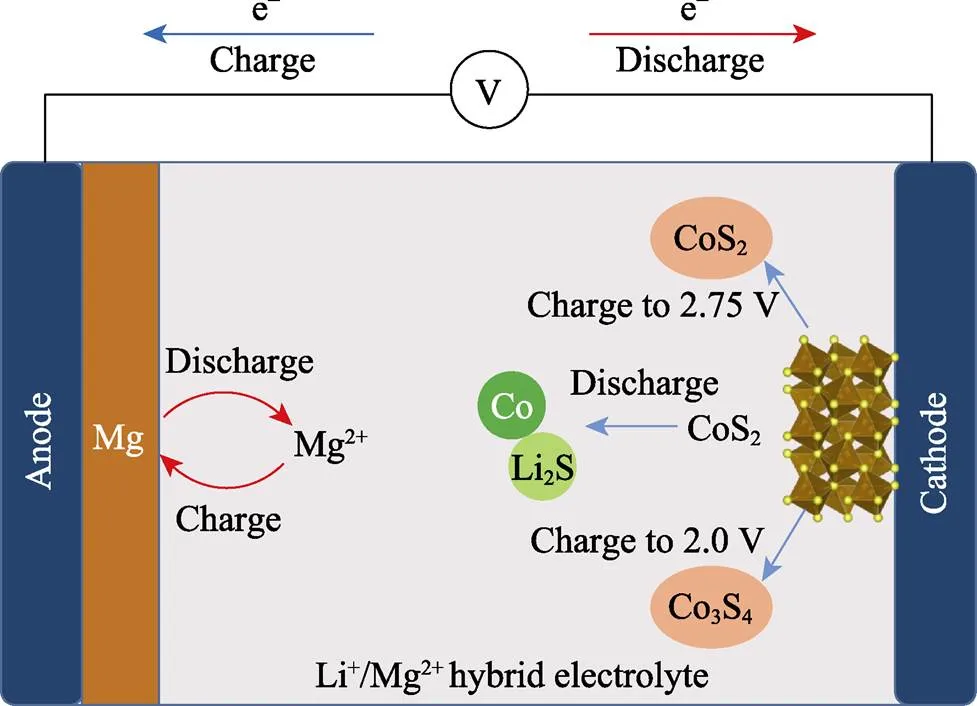
图6 基于锂镁混合电解液的Mg-CoS2电池在不同电压区间下的充放电过程示意图
当充电电位提高至2.75 V时, 正极侧的还原产物单质Co和Li2S则会经过电化学氧化生成CoS2, 可逆的转换反应极大改善了电池容量的循环稳定性。与低电位区间充放电类似, 在整个过程中, 镁负极仍发生可逆的镁金属沉积/剥离。电池在高电位区间首次充电的电化学反应可表述为:
首次充电至2.75 V:
Co0+2Li2S→CoS2+4Li++4e–(5)
Mg2++2e–→Mg (6)
由于CoS2正极在低充电电位下的氧化产物为Co3S4, 为此还测试了Co3S4作为初始正极在锂镁双盐电解液中的电化学性能。采用XRD对水热法合成的Co3S4进行物相分析, 结果如图S4所示。Co3S4样品的衍射峰与立方相Co3S4(PDF#47-1738)的标准衍射峰吻合, 没有出现明显的杂质峰, 且其衍射峰形尖锐, 代表合成产物的结晶度较好。在0.1~ 2.0 V充放电范围内, Co3S4正极在1 mol/L LiCl-APC电解液中的电化学性能如图S5所示。Mg-Co3S4电池的初始放电比容量仅有~226 mAh/g, 随后其放电比容量迅速衰减, 循环20圈后放电比容量仅有~10 mAh/g, 其充放电曲线表现出很大的电压极化。该结果进一步表明CoS2在低充电电位下不可逆生成的Co3S4是电池性能衰减的主要原因。
此外, 计算CoS2在不同电池体系中的热力学平衡电位, 用来比较对应电池的反应驱动力[35]。如图7所示, Mg-CoS2电池在锂镁双盐电解液中的放电反应(CoS2+2Mg+4Li+→Co0+2Li2S+2Mg2+)由Li+驱动, 其热力学平衡电位为1.25 V; 而在纯镁电解液中, 由Mg2+驱动的放电反应(CoS2+2Mg→Co0+2MgS)的热力学平衡电位为1.39 V (计算细节见补充材料)。这说明从热力学角度上, Mg2+驱动的转换反应应该更早发生, 但在实际过程中, 锂镁混合体系的正极反应由Li+主导驱动而非Mg2+。这应该是由于Mg2+电荷密度高, 在正极中扩散缓慢。受制于动力学因素, Mg2+驱动的反应在活性材料表面会受到限制, 从而导致CoS2正极在纯镁电解液中的性能很差。此外, 还比较了CoS2正极在锂镁混合体系(MLB)和锂金属电池(LMB)中的热力学平衡电位和实际放电平台电位间的差值。Mg-CoS2在锂镁混合电解液中的实际放电平台电压约为0.80 V, 要比热力学平衡电位低0.45 V; 而Li-CoS2电池的实际放电平台电压(1.63 V)仅比其热力学平衡电位(1.90 V)低0.27 V。Mg-CoS2电池的实际放电平台电压与热力学平衡值的电压偏差更大, 说明Mg2+的高电荷密度会引起更强的溶剂化作用, 从而提高负极侧Mg(solvent)2+/ Mg的氧化还原电位, 进而增大电压极化[15]。然而基于锂镁双盐电解液的Mg-CoS2电池在0.1~2.75 V电压区间的放电比容量却能与Li-CoS2电池相媲美, 进一步表明加入锂盐能促进电池的动力学。
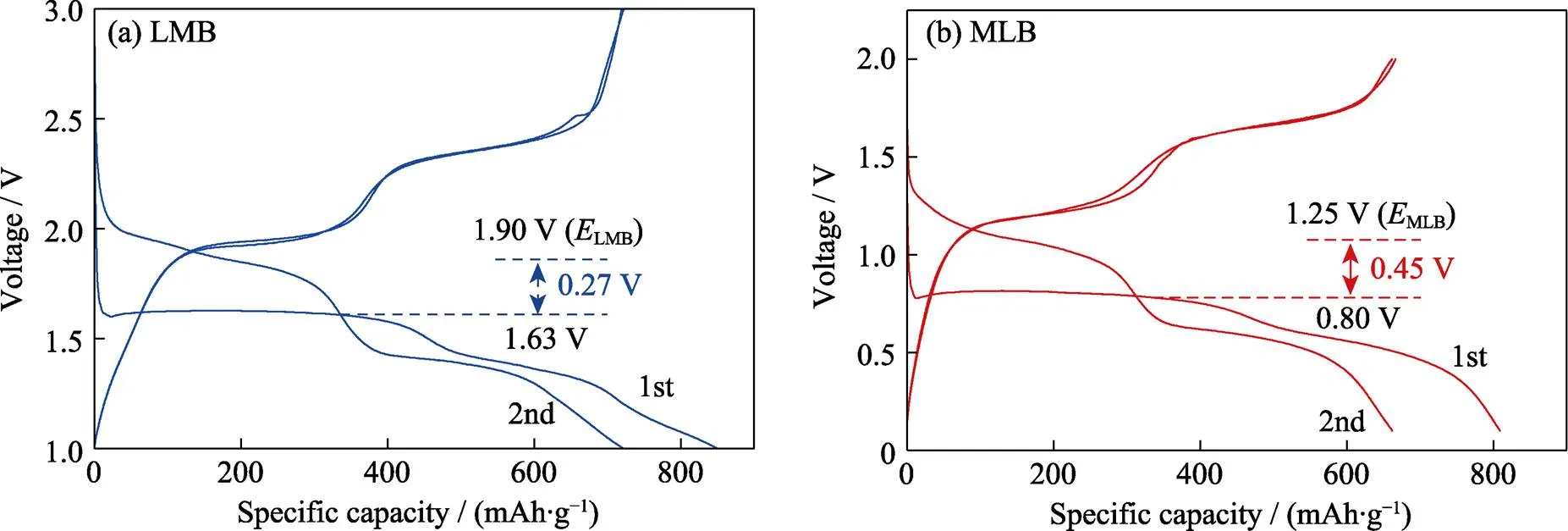
图7 (a)Li-CoS2(LMB)和(b)Mg-CoS2(MLB)电池的前2圈充放电曲线的热力学电位和实际放电电位的比较
众所周知, Mg2+的高电荷密度所引起的极化效应会降低正极的反应动力学, 在持续充放电过程中导致正极材料结构坍塌和循环稳定性快速衰退。本研究证明锂盐添加剂体系比单镁盐体系更有优势, 锂镁双盐体系中的Li+能代替Mg2+激活正极反应, 提高电池的容量。但过窄的充放电电位区间会导致CoS2正极发生不可逆反应, 导致电池循环稳定性显著降低, 而合适的充放电区间才能进一步保证双盐电解液对电池动力学的激活效果。尤其对于过渡金属硫化物, 其在充放电过程中产生的多硫化物会溶解在电解液中, 进而引起活性物质和对应电池容量的不可逆损失, 而适配的充放电电位能提高金属硫化物正极的转换反应可逆性和循环稳定性。
3 结论
综上所述, 本工作报道了锂镁双盐电解液和充放电电压区间对CoS2正极转换反应性能的影响。锂镁双盐电解液中的Li+替代Mg2+驱动正极反应, 可以克服Mg2+在固相中扩散缓慢的问题; 而且负极仍发生镁金属的可逆沉积/剥离, 在长期循环过程中不形成枝晶, 提高了负极的稳定性和安全性。在常见的0.1~2.0 V的充放电区间内, CoS2正极的容量在不同种类的锂镁混合电解液中均会迅速衰减, 且与锂盐的浓度无关。电池容量衰减与CoS2正极在低充电截止电位下的不可逆相变反应有关, 在首次充电过程中生成的是Co3S4而非CoS2, Co3S4在充放电过程中Co和S元素的溶解进一步加剧了容量的不可逆损失。而提高充电电位至2.75 V能显著改善CoS2正极的转换反应可逆性和循环稳定性, 双盐镁电池循环150圈后, 其放电比容量仍高达275 mAh/g, 且在1高倍率下放电比容量保持在~210 mAh/g。本工作证明采用锂盐添加剂策略和适配的充放电电位调控能促进过渡金属硫化物在转换反应型镁金属电池中的电化学性能, 为过渡金属硫化物或硒化物在镁电池中的发展提供了参考。
本文相关补充材料可登陆https://doi.org/10.15541/jim20210353查看。
[1] LIU B, ZHANG J G, XU W. Advancing lithium metal batteries., 2018, 2(5): 833–845.
[2] LI C, CHEN K, ZHOU X,. Electrochemically driven conversion reaction in fluoride electrodes for energy storage devices., 2018, 4(1): 22.
[3] LIN D, LIU Y, CUI Y. Reviving the lithium metal anode for high-energy batteries., 2017, 12(3): 194–206.
[4] ZHAI P, LIU L, GU X,. Interface engineering for lithium metal anodes in liquid electrolyte., 2020, 10(34): 2001257.
[5] YOO H D, SHTERENBERG I, GOFER Y,. Mg rechargeable batteries: an on-going challenge., 2013, 6(8): 2265–2279.
[6] ZHOU X, TIAN J, HU J,. High rate magnesium-sulfur battery with improved cyclability based on metal-organic framework derivative carbon host., 2018, 30(7): 1704166.
[7] MULDOON J, BUCUR C B, GREGORY T. Quest for nonaqueous multivalent secondary batteries: magnesium and beyond., 2014, 114(23): 11683–11720.
[8] AURBACH D, GIZBAR H, SCHECHTER A,. Electrolyte solutions for rechargeable magnesium batteries based on organomagnesium chloroaluminate complexes., 2002, 149(2): A115–A121.
[9] EAVES-RATHERT J, MOYER K, ZOHAIR M,. Kinetic-versus diffusion-driven three-dimensional growth in magnesium metal batteries., 2020, 4(6): 1324–1336.
[10] LU Z, SCHECHTER A, MOSHKOVICH M,. On the electrochemical behavior of magnesium electrodes in polar aprotic electrolyte solutions., 1999, 466(2): 203–217.
[11] SON S B, GAO T, HARVEY S P,. An artificial interphase enables reversible magnesium chemistry in carbonate electrolytes., 2018, 10: 532–539.
[12] BUCUR C B, GREGORY T, OLIVER A G,. Confession of a magnesium battery., 2015, 6(18): 3578–3591.
[13] LEVI E, LEVI M D, CHASID O,. A review on the problems of the solid state ions diffusion in cathodes for rechargeable Mg batteries., 2009, 22: 13–19.
[14] YAGI S, ICHITSUBO T, SHIRAI Y,. A concept of dual-salt polyvalent-metal storage battery., 2014, 2(4): 1144–1149.
[15] ZHANG Y, XIE J, HAN Y,. Dual-salt Mg-based batteries with conversion cathodes., 2015, 25(47): 7300–7308.
[16] WU N, YANG Z Z, YAO H R,. Improving the electrochemical performance of the Li4Ti5O12electrode in a rechargeable magnesium battery by lithium-magnesium co-intercalation., 2015, 127(19): 5849–5853.
[17] CHO J H, AYKOL M, KIM S,. Controlling the intercalation chemistry to design high-performance dual-salt hybrid rechargeable batteries., 2014, 136(46): 16116–16119.
[18] ZHANG Z, XU H, CUI Z,. High energy density hybrid Mg2+/Li+battery with superior ultra-low temperature performance., 2016, 4(6): 2277–2285.
[19] GAO T, HAN F, ZHU Y,. Hybrid Mg2+/Li+battery with long cycle life and high rate capability., 2015, 5(5): 1401507.
[20] SU S, NULI Y, HUANG Z,. A high-performance rechargeable Mg2+/Li+hybrid battery using one-dimensional mesoporous TiO2(B) nanoflakes as the cathode., 2016, 8(11): 7111–7117.
[21] PAN W, LIU X, MIAO X,. Molybdenum dioxide hollow microspheres for cathode material in rechargeable hybrid battery using magnesium anode., 2015, 19: 3347–3353.
[22] CHENG Y, CHANG H J, DONG H,. Rechargeable Mg-Li hybrid batteries: status and challenges., 2016, 31: 3125–3141.
[23] CHEN X, WANG S, WANG H. High performance hybrid Mg-Li ion batteries with conversion cathodes for low cost energy storage., 2018, 265: 175–183.
[24] WU C, HU J, TIAN J,. Stacking of tailored chalcogenide nanosheets around MoO2-C conductive stakes modulated by a hybrid POM⊂MOF precursor template: composite conversion- insertion cathodes for rechargeable Mg–Li dual-salt batteries., 2019, 11(6): 5966–5977.
[25] GAO T, NOKED M, PEARSE A J,. Enhancing the reversibility of Mg/S battery chemistry through Li+mediation., 2015, 137(38): 12388–12383.
[26] YUAN H, YANG Y, NULI Y,. A conductive selenized polyacrylonitrile cathode in nucleophilic Mg2+/Li+hybrid electrolytes for magnesium-selenium batteries., 2018, 6(35): 17075–17085.
[27] WANG Q, ZOU R, XIA W,. Facile synthesis of ultrasmall CoS2nanoparticles within thin N-doped porous carbon shell for high performance lithium-ion batteries., 2015, 11(21): 2511–2517.
[28] LIU L, KANKAM I, ZHUANG H L. Single-layer antiferromagnetic semiconductor CoS2with pentagonal structure., 2018, 98(20): 205425–205430.
[29] LIU W, HU E, JIANG H,. A highly active and stable hydrogen evolution catalyst based on pyrite-structured cobalt phosphosulfide., 2016, 7: 10771.
[30] MULDOON J, BUCUR C B, OLIVER A,. Corrosion of magnesium electrolytes: chlorides-the culprit., 2013, 6(2): 482–487.
[31] TIAN J, CAO D, ZHOU X,. High-capacity Mg-organic batteries based on nanostructured rhodizonate salts activated by Mg-Li dual-salt electrolyte., 2018, 12(4): 3424–3435.
[32] TAO M, DU G, YANG T,. MXene-derived three-dimensional carbon nanotube network encapsulate CoS2nanoparticles as an anode material for solid-state sodium-ion batteries., 2020, 8(6): 3018–3026.
[33] JIN H, WANG J, SU D,.cobalt-cobalt oxide/N-doped carbon hybrid as superior bifunctional electrocatalysts for hydrogen and oxygen evolution., 2015, 137(7): 2688–2694.
[34] GUO C, ZHANG W, LIU Y,. Constructing CoO/Co3S4heterostructures embedded in N-doped carbon frameworks for high-performance sodium-ion batteries., 2019, 29(29): 1901925.
[35] ZU C X, LI H. Thermodynamic analysis on energy densities of batteries., 2011, 4(8): 2614–2624.
补充材料:
双盐镁电池CoS正极材料的电化学性能研究
李文博1,2, 黄民松2,3, 李月明1, 李驰麟2,3,4
(1. 景德镇陶瓷大学 材料科学与工程学院, 景德镇 333403; 2. 中国科学院 上海硅酸盐研究所, 高性能陶瓷和超微结构国家重点实验室, 上海 201899; 3. 中国科学院大学 材料与光电研究中心, 北京 100049; 4. 中国科学院 上海硅酸盐研究所, 中国科学院能量转换材料重点实验室, 上海 201899)
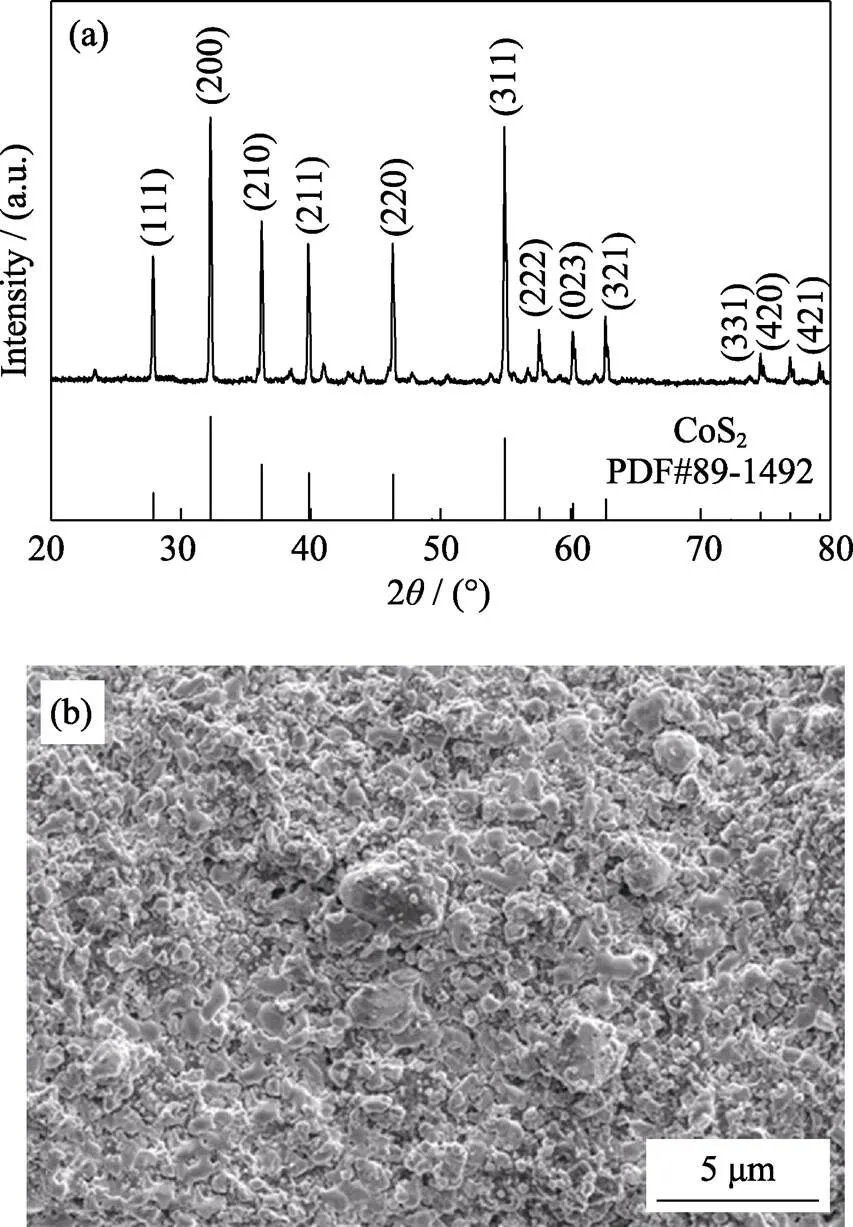
图S1 (a)商业CoS2的XRD图谱, (b) CoS2正极循环前的SEM照片
Fig. S1 (a) XRD pattern of commercially available CoS2,(b) SEM image of CoS2cathode before cycling

图S2 CoS2在纯APC电解液中的充放电曲线
Fig. S2 Discharge-charge curves of CoS2cathode in pure APC electrolyte

图S3 Li-CoS2电池的前20次充放电曲线
Fig. S3 Discharge-charge curves of Li-CoS2battery during the first 20 cycles
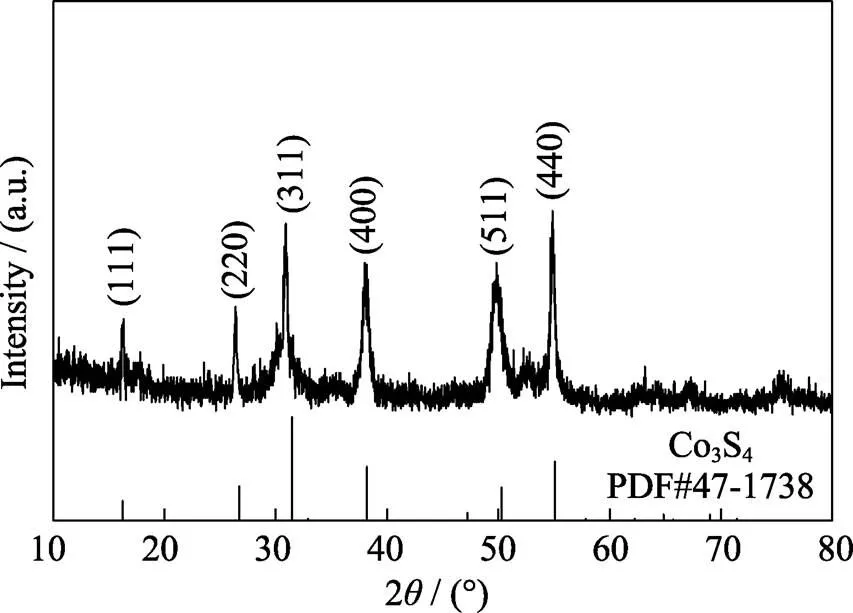
图S4 合成的Co3S4样品的XRD图谱
Fig. S4 XRD pattern of as-synthesized Co3S4sample
基于图7的讨论:
基于锂镁双盐电解液的Mg-CoS2电池(MLB) 的反应可表述为: CoS2+2Mg+4Li+→Co0+2Li2S+ 2Mg2+。其实际放电平台电压 (~0.80 V) 要小于热力学平衡电位MLB(1.25 V, 计算细节如下)。此外, Li-CoS2电池(LMB, 基于1 mol/L LiTFSI/DGM电解液) 的实际放电平台电压为1.63 V, 比通过放电反应CoS2+4Li→Co0+2Li2S计算得到的热力学平衡电位LMB(1.90 V)要低0.27 V。Mg-CoS2电池具有与热力学平衡值相比更大的电压偏差, 证明Mg2+的高电荷密度会引起更强的溶剂化作用, 从而提高负极侧Mg(solvent)2+/Mg的氧化还原电位, 进而增大电压极化。
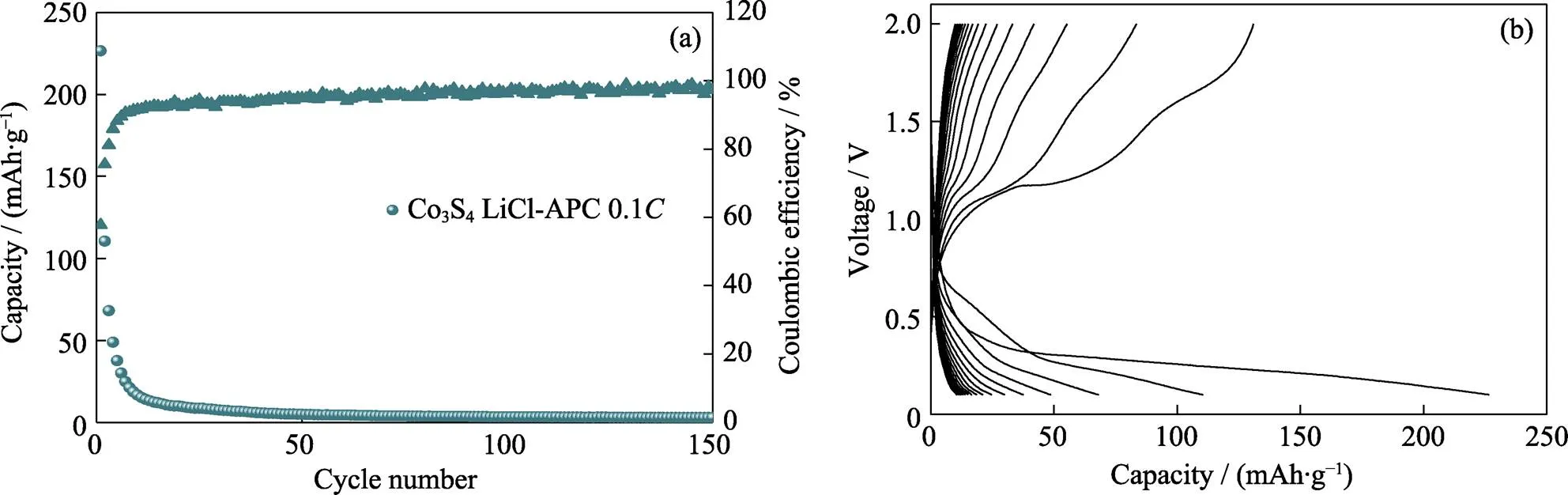
图S5 Co3S4正极在1 mol/L LiCl-APC电解液中的(a)长循环性能和(b)对应的充放电曲线
Fig. S5 (a) Long-term cycling performance and (b) corresponding discharge-charge curves of Co3S4cathode in 1 mol/L LiCl-APC electrolyte
1=0.91 mA/cm2
基于锂镁双盐电解液的Mg-CoS2电池的热力学平衡电位计算假设在室温且在1个标准大气压下进行电池测试, 电池的放电反应为: CoS2+2Mg+ 4Li+→ Co0+2Li2S+2Mg2+, 则基于上述放电反应的总标准吉布斯自由能可表示为:

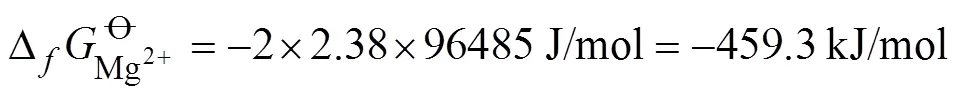
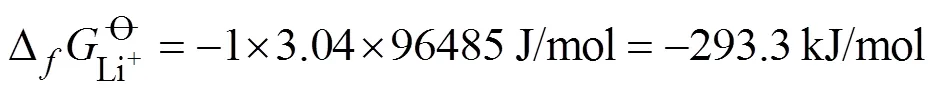
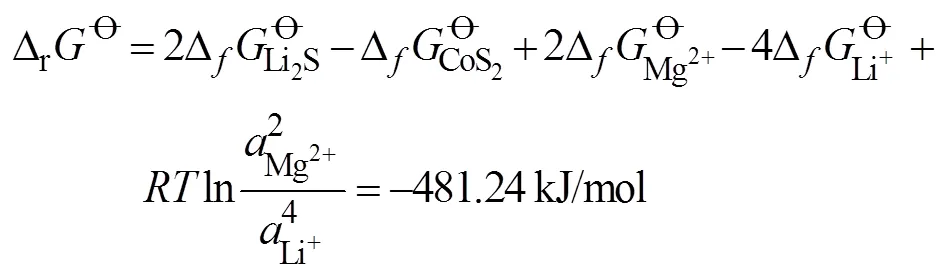

类似的, 可通过上述方法计算得到Li-CoS2和Mg-CoS2在单盐电解液中的热力学平衡电位为1.90和1.39 V。两者的放电反应可表示为:
CoS2+ 4Li → Co0+ 2Li2S (S6)
CoS2+ 2Mg → Co0+ 2MgS (S7)
[1] ZU C X, LI H. Thermodynamic analysis on energy densities of batteries., 2011, 4(8): 2614–2624.
CoS2as Cathode Material for Magnesium Batteries with Dual-salt Electrolytes
LI Wenbo1,2, HUANG Minsong2,3, LI Yueming1, LI Chilin2,3,4
(1. School of Materials Science and Engineering, Jingdezhen Ceramic University, Jingdezhen 333403, China; 2. State Key Laboratory of High Performance Ceramics and Superfine Microstructure, Shanghai Institute of Ceramics, Chinese Academy of Sciences, Shanghai 201899, China; 3. Center of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China; 4. CAS Key Laboratory of Materials for Energy Conversion, Shanghai Institute of Ceramics, Chinese Academy of Sciences, Shanghai 201899, China)
Magnesium metal batteries (MMBs) have attracted increasing attention due to the high volume specific capacity (3833 mAh/cm3) and high safety of Mg metal anode. Nevertheless, the high polarization effect induced by Mg2+inhibits its diffusion in solid phase and therefore limits the specific capacity of MMBs. Li+/Mg2+dual-salt electrolyte has been proposed to circumvent the sluggish diffusion of Mg2+in solid phase, which enables Li+to replace Mg2+to drive the cathode reaction. In this work, the electrochemical performance of CoS2as conversion cathode of MMBs is studied based on different Li+/Mg2+dual-salt electrolytes, and the effects of Li-salt concentration and discharge-charge voltage range on cycling stability are analyzed. The strategy of Li-salt additive remarkably promotes the conversion kinetics of CoS2based MMBs. By developing charge voltage to 2.75 V, the cycling stability of Mg-CoS2cell in LiCl-APC electrolyte is significantly enhanced. Its specific capacity can be maintained at 275 mAh/g after 150 cycles, which is much higher than that (33 mAh/g) under the protocol of 2.0 V charge voltage. It is found that the capacity degradation of MMBs is related to the irreversible reaction of CoS2and generation of Co3S4at the charge potential of 2.0 V. And the dissolution of Co and S elements from active species aggravates the irreversible loss of capacity. This study provides an electrochemical activation solution to the development of transition metal sulfides in conversion type MMBs.
magnesium metal battery; CoS2; Li/Mg dual-salt system; cathode material; conversion reaction
1000-324X(2022)02-0173-09
10.15541/jim20210353
O614
A
2021-06-04;
2021-08-11;
2021-08-20
国家自然科学基金 (U1830113, 51772313, 21975276); 国家重点研发计划(2016YFB0901600)
National Natural Science Foundation of China (U1830113, 51772313, 21975276); National Key R&D Program of China (2016YFB0901600)
李文博(1996–), 男, 硕士研究生. E-mail: 1920024021@stu.jci.edu.cn
LI Wenbo(1996–), male, Master candidate. E-mail: 1920024021@stu.jci.edu.cn
李月明, 教授. E-mail: lym6329@163.com; 李驰麟, 研究员. E-mail: chilinli@mail.sic.ac.cn
LI Yueming, professor. E-mail: lym6329@163.com; LI Chilin, professor. E-mail: chilinli@mail.sic.ac.cn
