一种靶向Wee1蛋白激酶的小分子抑制剂体外筛选模型的建立
2022-04-12周秋华苗雷季晓君左锐吴舰徐丹
周秋华,苗雷,季晓君,左锐,吴舰,徐丹
肿瘤发生的三大基础条件是细胞周期失控、DNA 损伤修复(DNA damage repair,DDR)和肿瘤免疫逃逸。在细胞周期进程中,细胞周期检查点发挥着重要作用,通过细胞周期检查点(如 G1/S,G2/M)检测 DNA 是否受到损伤,进而决定细胞周期是否进入下一个环节。由于遗传畸变经常发生在G1/S 检查点,因此肿瘤细胞更加依赖 S/G2和G2/M 检查点来维持基因组的完整性。p53 蛋白参与细胞周期的调控和 DDR,通过 ATM/Chk2-p53途径在 G1/S 期和 G2/M 期监测基因组的完整性[1]。在恶性肿瘤中,50% 以上的肿瘤存在 p53 基因缺陷或突变,导致细胞周期 G1/S 检查点的缺陷,使得肿瘤细胞 DNA 的复制及损伤修复过程更依赖于 G2/M 检查点[2],防止有丝分裂过程中产生大量的 DNA 损伤导致细胞凋亡。在正常细胞中,G1/S检查点未受到损伤,因此,G2/M 检查点不会承受在 DNA 损伤修复之前停止细胞周期的负担。这支持了废除 G2/M 检查点将选择性地影响肿瘤细胞,而不影响正常的细胞生长的假说。
Wee1 激酶是丝氨酸/苏氨酸蛋白激酶家族中的一员,它的功能是在细胞出现 DNA 损伤时,参与细胞周期 G2/M 检查点对 DNA 损伤检查点的正调控,从而抑制细胞进入有丝分裂期[3]。Wee1 激酶在 DNA 损伤应答通路中起到了关键作用,当细胞的 DNA 损伤被识别后,Wee1 被磷酸化激活,进而磷酸化失活 Cdc2(Thr15),使细胞周期停顿,暂不进入有丝分裂,允许细胞有足够的时间进行DNA 损伤修复。p53 基因缺陷的癌细胞更依赖于Wee1 来维持细胞正常的有丝分裂进程[4]。而在p53 基因功能正常的细胞中,抑制 Wee1 激酶的活性,可以通过 p53 依赖的方式进行 DNA 损伤修复,正常细胞的生存不受影响[5]。因此,对于 p53 基因缺陷肿瘤细胞,Wee1 抑制剂可以作为一种有效的结合 DNA 损伤治疗的致敏剂,使得细胞过早进入有丝分裂以及在有丝分裂期间的细胞死亡[4]。因此,Wee1 激酶是通过 DNA 损伤修复机制发挥抗肿瘤作用的极具潜力的药物开发靶点。
目前,在研的 Wee1 小分子抑制剂主要有PD0166285、PD0407824 以及 MK-1775 等。MK-1775(又名 AZD-1775 或 adavosertib)是阿斯利康公司研发的一款高选择性的 Wee1 抑制剂。在包含 223 个激酶的激酶谱研究中,1.0 μmol/L 的MK-1775 只对其中 8 个激酶的抑制率超过 80%。对其中 7 个激酶的活性比 Wee1 激酶(IC50=5 nmol/L)低 10 倍,对 MYT1 激酶的活性比 Wee1激酶低 100 倍。将 MK-1775 与不同作用机制的DNA 损伤剂联用,包括抗代谢物(卡培他滨、5-氟尿嘧啶、吉西他滨和培美曲塞)、DNA 交联剂(卡铂、顺铂和丝裂霉素 C)或拓扑异构酶抑制剂(喜树碱和多柔比星),在 p53 缺失或突变的宫颈癌、结肠癌、肺癌和胰腺癌细胞系中显示了增强的抗肿瘤疗效,这些结果支持有丝分裂致死性的作用机制[6]。MK-1775 是目前临床进展最快,研究最多的 Wee1 抑制剂。I 期临床结果显示,MK-1775 单药治疗耐受性良好,剂量爬坡未达到最大耐受剂量。在与化疗药物联合治疗的人群中,不良反应主要包括疲劳、恶心和呕吐、腹泻和血液学毒性[7]。II 期临床研究证实,MK-1775 可使癌症患者对不同的化疗药物致敏。在 p53 突变的铂类药物难治或耐药的卵巢癌患者中,MK-1775 联合卡铂治疗均显示出了一定的疗效[8]。
在本研究中,我们建立了一种新颖、有效的Wee1 激酶小分子抑制剂体外筛选模型。为了获得单药细胞毒性较低的 Wee1 选择性抑制剂,先通过酶学筛选出对 Wee1 激酶有抑制活性的小分子,后在 WiDr 细胞中进行单用及与吉西他滨联用的细胞增殖抑制评价,同时通过测定细胞内 Wee1 下游Cdc2 及 HH3 的磷酸化水平综合评估 Wee1 抑制剂的活性及细胞毒性作用。本文以 MK-1775 为例进行综合活性评价分析。
1 材料与方法
1.1 材料
1.1.1 试剂 Wee1 激酶购自日本 Carna biosciences 公司;ADP-GloTMKinase Assay Kit 购自美国 Promega 公司;MK-1775、吉西他滨购自上海毕得医药公司;MEM 培养基、胎牛血清(FBS)、NEAA 非必需氨基酸、GlutaMAX 以及丙酮酸钠均购自美国 Gibco 公司;DMSO 购自美国 Sigma 公司;Cell Counting-LiteTM2.0 溶液购自南京诺唯赞生物科技有限公司;Phospho-Cdc2(Tyr15) (10A11) 兔 mAb、Cdc2 (POH1) 鼠 mAb以及 GAPDH (D4C6R) 鼠 mAb 均购自美国 CST公司;Phospho-Histone H3 (Ser10) 兔抗体购自美国Millipore 公司;IRDye 800CW 羊抗兔 IgG (H +L)、RDye 680RD 羊抗鼠 IgG (H + L) 以及 Li-Cor封闭缓冲液均购自美国 Li-COR 公司。
1.1.2 仪器 自动加液仪 D300e 以及多功能酶标仪购自瑞士 Tecan 公司;自动洗板分液仪购自美国 Bio Tek 公司;生化培养箱、CO2培养箱以及荧光倒置显微镜购自美国 Thermo 公司;荧光细胞计数仪购自艾力特生命科学(上海)有限公司;生物安全柜购自苏净安泰公司;Apricot 移液工作站购自美国 Apricot Designs 公司;Odyssey CLx 双色红外激光成像系统购自美国 Li-COR 公司。
1.1.3 细胞 p53 突变型人结肠癌细胞(WiDr 细胞)购自南京科佰生物科技有限公司,根据供应商指示的培养条件(含 10% FBS、1% 谷氨酰胺、1%NEAA 和 1% 丙酮酸钠的 MEM 培养基)进行培养。
1.2 方法
1.2.1 体外激酶实验 重组激酶 Wee1 在含40 mmol/L Tris-HCl、20 mmol/L MgCl2、0.1 mg/ml BSA、2 mmol/L DTT 的激酶缓冲液(pH 7.5)中调整反应浓度为 0.67 mg/L。然后采用自动加液仪加入MK-1775 化合物,起始浓度 1 μmol/L,1/2 Log 倍梯度稀释,共设 8 个梯度,在 30 ℃ 条件下孵育15 min。接着与 2 μg/ml 的底物及 50 μmol/L 的ATP 在 30 ℃ 反应 120 min,最后采用 ADP-Glo法评价目标化合物抑制 Wee1 激酶的活性。实验设置 3 次重复。
1.2.2 细胞增殖抑制实验 取适量的 WiDr 细胞株接种于白壁底透的 96 孔板中,细胞贴壁 24 h后,用自动加液仪加入吉西他滨进行损伤作用,摸索吉西他滨合适的损伤时间及浓度,然后用自动加液仪加入梯度浓度的 MK-1775 进行联用,摸索合适的联用时间。联用组同时与单药吉西他滨以及单药 MK-1775 比较对细胞活性的影响。细胞活力采用 Cell Counting-LiteTM2.0 溶液评价,采用多功能酶标仪测定 luminescence 信号。
1.2.3 Odyssey in-cell Western 验证 取适量的WiDr 细胞株接种于 384 孔板中,细胞贴壁过夜后,用 10 nmol/L 的吉西他滨处理 WiDr 细胞,37 ℃、5% CO2孵育 24 h。第 3 天加入化合物MK-1775(DMSO 最终浓度为 0.1%),37 ℃、5%CO2孵育 7 h。然后加入 8% 固定液固定细胞,100% 甲醇渗透细胞。PBS 洗涤后用 Li-Cor 封闭缓冲液封闭。去除封闭缓冲液,加入一定比例的一抗混合液(兔抗-pCdc2/鼠抗-Cdc2 或兔抗-pHH3/鼠抗-GAPDH),4 ℃ 孵育过夜。用 PBST(0.05%Tween 20 的 PBS)洗涤板子 3 次后加入一定比例的二抗混合液(羊抗兔 800CW/羊抗鼠 680RD),避光孵育 45 min。用 PBST洗涤板子 3 次。以1000 r/min 倒置离心 1 min,用近红外双色荧光成像系统检测。
1.2.4 数据统计分析
1.2.4.1 体外激酶实验 所有数据分析均采用GraphPad Prism 5 软件拟合量效曲线,得到化合物对 Wee1 激酶抑制的 IC50值。实验重复 3 次,每次实验均设置单孔。抑制率计算公式:

其中,Lsample为样品孔的 luminescence 值;Lmin为无酶无待测化合物的空白对照孔的luminescence 均值;Lmax为有酶、无化合物的阴性对照孔的 luminescence 均值。
1.2.4.2 细胞增殖抑制实验 采用 GraphPad Prism 5 软件拟合量效曲线,得到化合物对细胞增殖抑制的 IC50值。实验重复 3 次,每次实验均设置复孔。抑制率计算公式:
抑制率(%)={[1 -(受试物信号值 - 空白组信号值)]/(阴性对照组信号值 - 空白组信号值)}× 100%
其中,受试物信号值为细胞 + 培养基 + 化合物组荧光信号均值;空白组信号值为培养基组荧光信号均值;阴性对照组信号值为细胞 + 培养基组荧光信号均值。
1.2.4.3 Odyssey in-cell Western 验证 采用GraphPad Prism 5 软件拟合量效曲线,得到化合物对 pCdc2 和 pHH3 的 IC50和 EC50值。每次实验均设置复孔。
2 结果
2.1 体外 Wee1 激酶抑制试验
MK-1775 是一种有效的选择性 Wee1 抑制剂,在体外激酶活性检测中 3 次实验的 IC50值分别为 1.990、2.771 和 3.281 nmol/L,平均 IC50值为 2.681 nmol/L,CV 值为 24%(图1)。通过该实验方法可以筛选出对 Wee1 激酶抑制活性较高的化合物。

图1 MK-1775 对 Wee1 激酶的抑制作用Figure 1 Inhibition of Wee1 kinase by MK-1775
2.2 体外单药与联用对 WiDr 细胞株的增殖抑制
2.2.1 吉西他滨损伤条件的优化 参考相关文献[9]摸索吉西他滨的损伤作用时间。吉西他滨作用48 h 后对 WiDr 细胞的损伤作用较弱,10 μmol/L最大抑制率不到 20%(图2A)。当作用时间延长到 72 h后,10 μmol/L 的吉西他滨最大抑制率可达到 60%,损伤作用比作用 48 h 有大幅增强(图2B)。当吉西他滨与不同浓度的 MK-1775 联用时,MK-1775 明显增强了吉西他滨对 WiDr 细胞的增殖抑制作用,且抑制作用呈现明显的浓度依赖性(图2C)。根据吉西他滨作用 72 h 的结果,选择了 1.5、4.5、13.5 nmol/L 的吉西他滨单药抑制率分别在 14%、28%、40% 的条件下观察其与MK-1775 的联用作用。
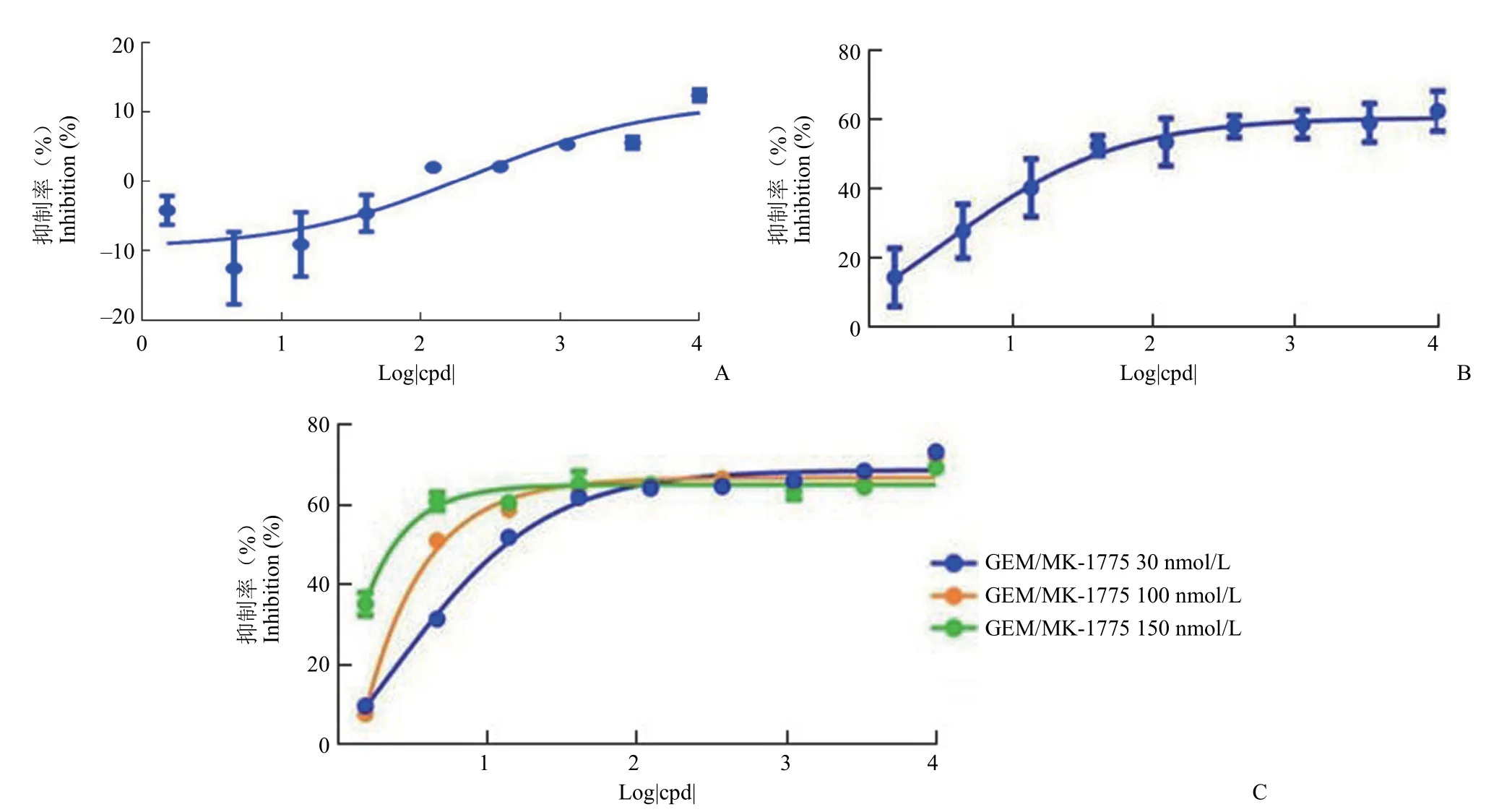
图2 WiDr 细胞中吉西他滨作用时间的优化(A:48 h;B:72 h;C:吉西他滨与 MK-1775 联用)Figure 2 Optimization of gemcitabine action time in WiDr cells (A: 48 h; B: 72 h; C: Gemcitabine incubated with MK-1775)
2.2.2 吉西他滨损伤浓度以及 MK-1775 作用时间的优化 将 3 个不同浓度的吉西他滨作用于 WiDr细胞 48 h 后,联合 MK-1775 分别作用 24、48 以及 72 h。结果显示,吉西他滨浓度为 1.5 nmol/L时,对 MK-1775 的增敏作用微弱,与 MK-1775的单药抑制率曲线几乎重合。吉西他滨浓度为13.5 nmol/L 时,对 MK-1775 的增敏作用虽然较强,但是抑制窗口较窄,这可能是由于吉西他滨浓度过高导致的细胞毒性作用,因此 4.5 nmol/L 是吉西他滨 3 个浓度中最为适宜的损伤浓度(图3)。从 MK-1775 化合物的作用时间来看,作用 72 h的药效窗口最大。因此,我们选择了 4.5 nmol/L 的吉西他滨损伤 48 h 后联用 MK-1775 化合物作用72 h 的条件用于后期在细胞水平评价 MK-1775的活性。
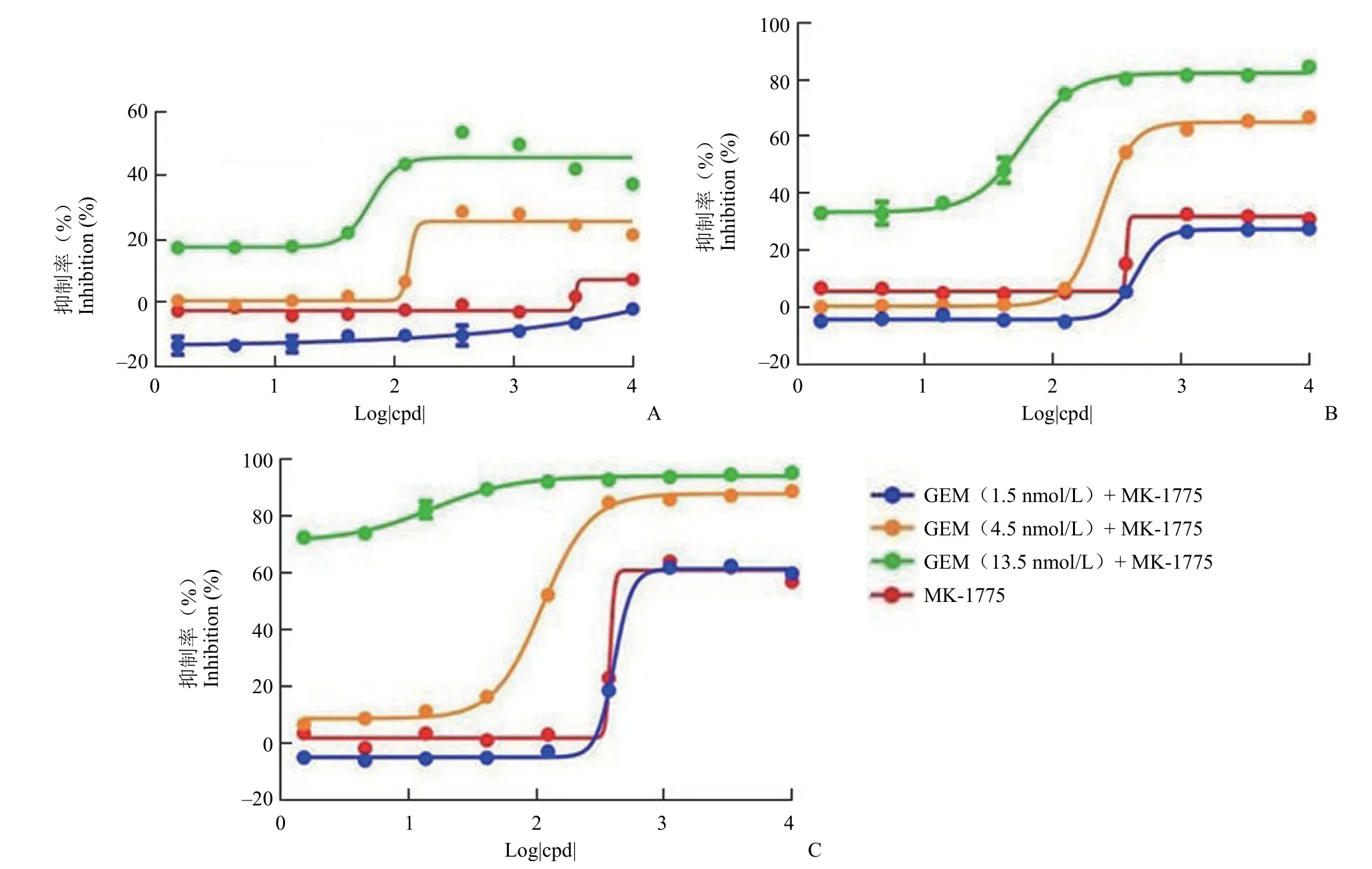
图3 WiDr 细胞中吉西他滨损伤浓度以及 MK-1775 作用时间的优化(A:联用 24 h;B:联用 48 h;C:联用 72 h)Figure 3 Optimization of gemcitabine damage concentration and time of MK-1775 in WiDr cells (A: 24 h; B: 48 h; C: 72 h)
2.2.3 WiDr 细胞增殖抑制筛选模型的应用 结果显示,MK-1775 单药对 WiDr 细胞的 IC50值分别为 380.2、488.1 和 352.2 nmol/L,3 次实验的CV 值为 18%,符合重复性的要求(图4A)。当MK-1775 与吉西他滨联用时,对 WiDr 细胞的IC50值分别为 114.0、155.6 和 85.0 nmol/L,3 次实验的 CV 值为 30%,表明该检测体系较为稳定(图4B)。3 次单药与联用实验 IC50的比值分别为 3.3、3.1、4.1,体现出了较为明显的联用效果。

图4 MK-1775 单用(A)及与吉西他滨联用(B)对 WiDr 细胞的增殖抑制作用Figure 4 Proliferation inhibition of WiDr cells by MK-1775 alone (A) or in combination with gemcitabine (B)
上述数据表明 DNA 损伤剂吉西他滨激活DNA 损伤通路后,Wee1 抑制剂加剧细胞毒性作用。4.5 nmol/L 的吉西他滨单用对细胞的抑制率仅为 15% 左右,但与 MK-1775 联用对细胞的增殖抑制作用显著增强。而 MK-1775 单用表现出温和的增殖抑制作用。通过单药的细胞增殖抑制筛选模型找到毒性较低的 Wee1 抑制剂,与吉西他滨损伤剂联用筛选出对损伤剂具有较强增敏作用的目标化合物。因此,该模型具有在早期细胞筛选预测单药 Wee1 抑制剂非靶标毒性,减少后期联用策略可能会产生的不良反应。与吉西他滨联用效果也为体内药效剂量的选择提供了参考。
2.3 p-Cdc2 和 pHH3 测定结果
吉西他滨处理后,MK-1775 对 WiDr 细胞中Wee1 下游底物 Cdc2(Tyr15)的磷酸化以及有丝分裂进程标志物组蛋白 H3(Ser10)的磷酸化可以反映 Wee1 激酶的活性。结果显示,MK-1775能够抑制 WiDr 细胞中 Cdc2 的磷酸化(IC50=157.2 nmol/L,图5A),使得由吉西他滨诱导的DNA 损伤检查点失效,并且通过诱导组蛋白 H3磷酸化(EC50= 103.4 nmol/L,图5B)使细胞过早进入有丝分裂,导致癌细胞死亡。这些结果可以预测化合物通过抑制 Wee1 激酶增强抗肿瘤作用。在激酶实验测定中有活性的化合物如果在细胞中是无活性的,可能是由于细胞渗透不足、培养基结合的差异或对生化测定读数的干扰。

图5 MK-1775 对 WiDr 细胞中 Cdc2(A)以及 HH3(B)磷酸化水平的影响Figure 5 Effects of MK-1775 on Cdc2 (A) and HH3 (B) phosphorylation in WiDr cells
3 讨论
DDR 是维持基因组稳定性的基本机制。有一些肿瘤细胞通过 DDR 通路的过表达或不受控制的激活已被证明可以保护癌细胞免受 DNA 损伤剂的杀伤作用。这些癌细胞能够耐受高增殖率诱导的复制应激,并维持内在的遗传不稳定性。Wee1 激酶在非恶性细胞的细胞周期中发挥调控遗传稳定性,而在癌细胞中具有促进 DNA 损伤修复和细胞周期控制的能力。多项研究表明,恶性细胞具有高表达的 Wee1,已成为化疗/放疗方案良好的预后生物标志物,癌细胞似乎严格依赖于 Wee1 激酶的功能来生存,特别是那些 p53 基因缺陷型肿瘤[8]。p53基因缺陷的各种恶性肿瘤使用传统标准的化疗/放疗疗法相比 p53 基因野生型的肿瘤细胞更为难治。截至目前,直接靶向 p53 的药物尚处于研发阶段,而合成致死的方法给予研究者们新的希望。Wee1 抑制剂联合放化疗疗法就是基于其原理,主要机制是 Wee1 抑制剂能够阻滞 p53 基因缺陷型肿瘤在 G2/M 期检查点的 DNA 损伤修复,使细胞过早进入有丝分裂从而导致细胞死亡。临床前和临床数据显示,Wee1 抑制剂 MK-1775 能增加 p53缺陷的癌细胞对放化疗的敏感性[10-12]。
MK-1775 已开展了大量的临床前研究,评价了其在单药和联合治疗中的疗效。关于其作用机制,当用于单药治疗时,MK-1775 能够诱导 S和(或)G2/M 细胞周期检查点跳跃。在联合治疗中,MK-1775 能够通过诱导细胞周期检查点跳跃、抑制 DNA 损伤修复和诱导凋亡来增强化疗(放疗)药物的细胞毒性[8]。体外实验表明,MK-1775 单用在 p53 野生型、p53 突变型和 p53 缺失型的肉瘤细胞中均观察到细胞毒性。MK-1775 单用产生的细胞毒性与其对 Wee1 的抑制无关,可能是其通过对 Cdc2(Tyr15)直接的磷酸化抑制导致肉瘤细胞过早进入有丝分裂或者由于其本身对 DNA 的损伤作用导致的细胞死亡[13]。考虑到 Wee1 抑制剂通常与化疗药物联合使用,上述情况可能会增加细胞毒性相关的不良事件,并破坏靶向 Wee1 的化学致敏策略[14]。事实上,目前还没有开发出针对其功能的选择性抑制剂。因此,研发单用毒性低的 Wee1抑制药物具有更加广阔的临床应用前景。
在本文中,我们描述了 MK-1775 的体外活性,它是一种有效的 Wee1 小分子抑制剂,与吉西他滨联用,在 p53 缺陷人结肠癌细胞中通过抑制细胞中 Cdc2 的磷酸化,导致 DNA 损伤检查点失效,通过磷酸化组蛋白 H3 诱导细胞过早进入有丝分裂,最终导致细胞死亡。但是 MK-1775 单药也存在一定的剂量依赖毒性。现有的 Wee1 抑制剂的细胞活性通常伴随着对有着相似结合位点的其他激酶的抑制,会带来脱靶作用。在早期筛选,从时间、成本、人力的角度我们不可能基于激酶谱筛选方法筛选靶向性更好的选择性抑制剂。我们采用单药增殖抑制实验和与损伤剂联用实验,旨在筛选出单药低毒并且对 DNA 损伤剂有增效作用的 Wee1抑制剂。该方法操作简单、方法稳定、评价成本较激酶谱筛选低,可在早期预测 Wee1 抑制剂脱靶细胞毒效应。该模型优化的关键因素为损伤剂作用的浓度与时间以及 Wee1 抑制剂作用的时间。这种筛选方法的意义在于避免单药的细胞毒作用,同时保证临床疗效,以改善安全性,可能使传统治疗不良预后的肿瘤对常规治疗更加敏感。
