团簇的密度泛函研究
2022-04-06胡纪平
胡纪平
(北京信息科技大学 理学院,北京 100192)
0 引 言
如何发现、改进、乃至于创造新材料、新物质,已成为当前应用领域和科技领域面临的严峻挑战。团簇作为存在于原子、分子与宏观固体物质之间的物质结构的新层次,具有其物理和化学性质随尺度不同而改变的特性,引起科研人员在理论和实验两方面的研究兴趣[1-4]。

1 理论计算
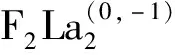
为了证明本文所采用的基组和赝势的正确性,在此计算了双原子分子体系的键长并与实验值比较,计算得到的F2的键长为0.140 79 nm,其实验值[16]为0.141 7±0.000 1 nm,La2的计算在前期工作已完成,与实验结果十分吻合,计算得到双原子FLa的键长、振动频率和结合能分别为0.201 8 nm、589 cm-1和6.84 eV,而曲晓燕等[17]给出的实验值分别为0.202 7 nm、570 cm-1和6.23 eV,由此可见采用的B3LYP方法及相应的基组和赝势用来研究含F和La元素的团簇是可行的,计算得到的数据是可靠的。
2 结果与讨论
计算得到的中性体系F2La2的稳定结构见图1,几何参数和能量见表1。
由图1和表1可见,优化得到的中性团簇的稳定结构共10个,直线型1个,具有D∞H对称性,自旋多重度为3,即3重态的直线结构(图1I)。平面结构有7个(图1II-VIII),其中3个具有D2H对称性的菱形结构(图1II-IV),结构II和III都具有F1-F2之间的键长略大于La1-La2键长的特点,结构II的自旋多重度1和3均优化出稳定结构,单重态的能量略低于三重态的能量,结构III只有自旋多重度为3的稳定结构,图1中的菱形结构IV自旋多重度为1,F1-F2之间的键长略小于La1-La2的键长;结构V为具有Cs对称性的平面结构,自旋多重度1和3均存在,单态的能量略高于三重态的能量;结构VI和VII是两个具有C2V对称性的梯形结构,单态和三重态均优化出来;结构VIII是具有C2V对称性的Y型结构,自旋多重度1和3均稳定存在。图1中的结构IX和X为立体结构,均具有C2V对称性,结构IX只优化出自旋多重度为1的构型,结构X两种自旋多重度1和3均存在。由能量最低原理可确定出体系的基态结构,由表1中10种结构的能量可判断其稳定性按照由强到弱的顺序为:III(3)>V(3)> V(1)>X(1)>IV(1)>X(3)>II(1)>II(3)>I(3)>VIII(3)>VI(1)>VI(3)>IX(1)>VIII(1)>VII(3)> VII(1),括号中的数字代表自旋多重度。由此可见3重态的菱形结构(图1III)为该体系的基态结构,键长F1-La1、F1-F2和La1-La2分别为0.233 7 nm、0.365 8 nm和0.291 0 nm,键角La1-F1-La2为77°,体系能量为-1 071.155 327 2 a.u.。

图1 F2La2团簇的稳定结构

表1 F2La2团簇稳定结构的几何参数
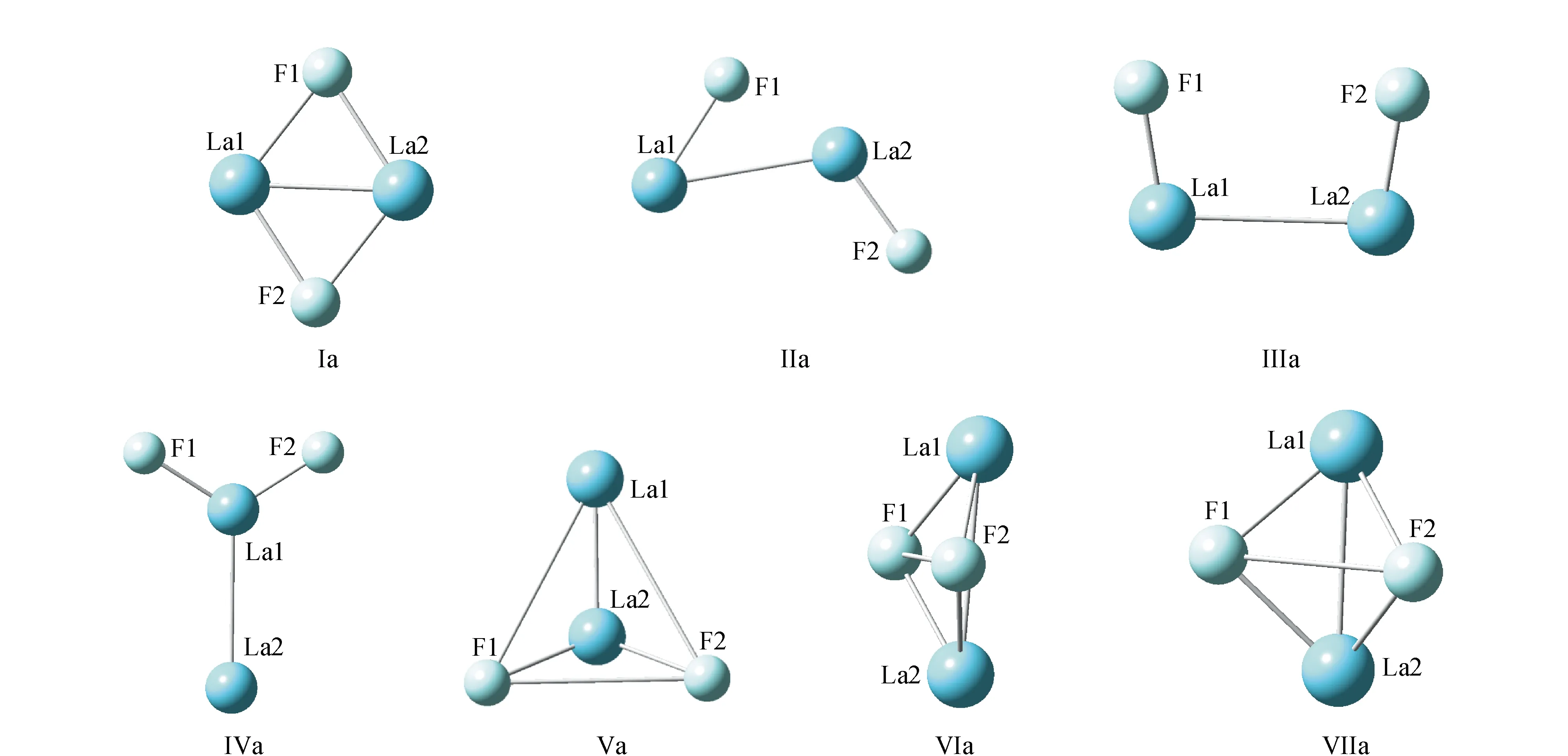
优化得到的负离子体系F2La2-1团簇的稳定结构见图2,几何参数和能量列于表2。
图团簇的稳定结构
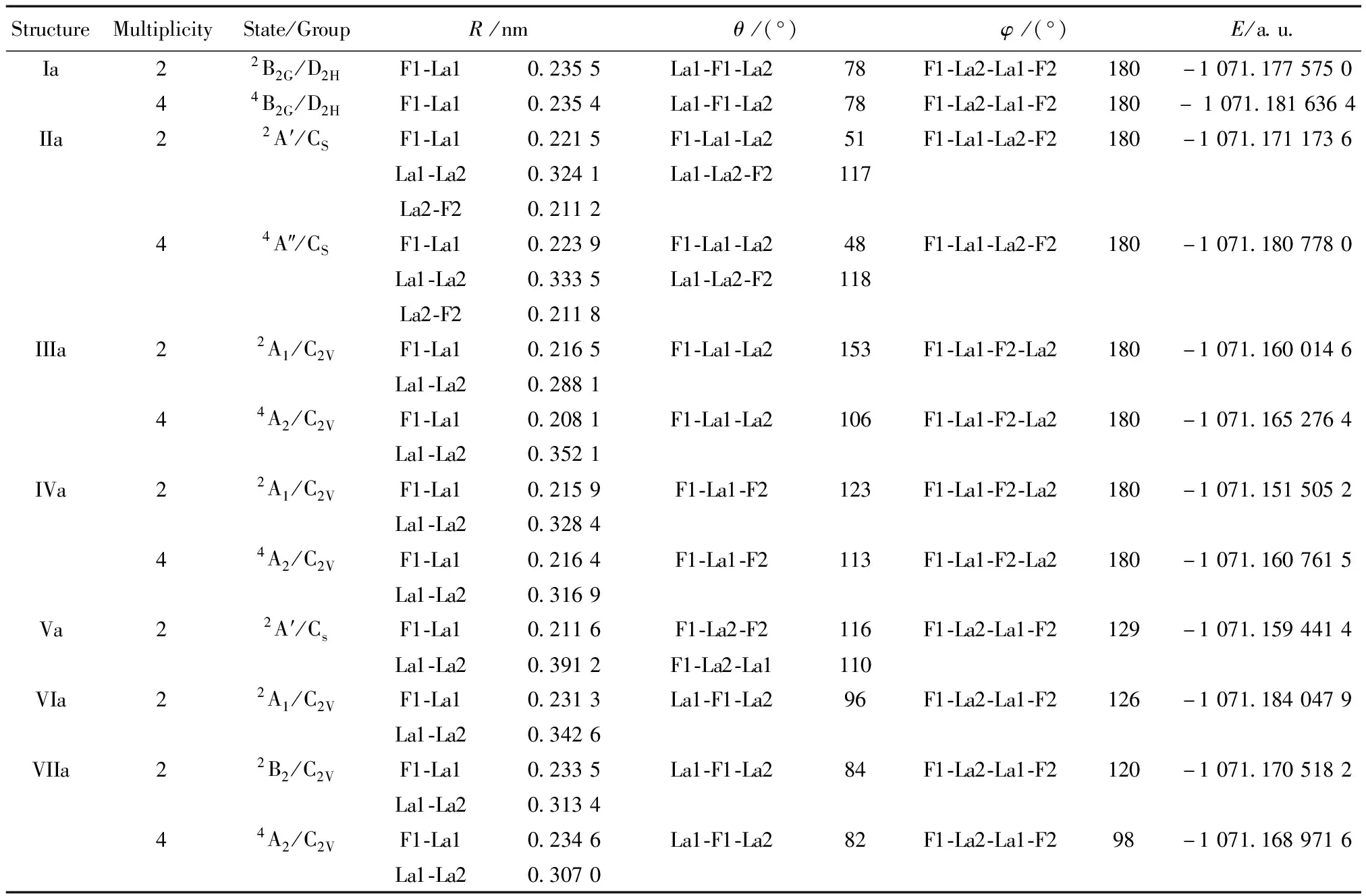
表2 F2La2-1团簇稳定结构的几何参数
由图2和表2可见,负一价体系的稳定结构有7个,其中平面结构4个,结构Ia至IVa分别为具有D2H对称性的菱形结构、CS对称性的平面结构、C2V对称性的梯形结构和C2V对称性的Y型结构,其中各种构型的四重态的能量均低于二重态的能量;立体结构有3个,Va至VIIa,Va具有CS对称性的二重态的立体结构,VIa和VIIa均是具有C2V对称性的蝴蝶结构,VIa只存在二重态,VIIa结构二重态和四重态均存在,四重态能量略高。按能量的大小得到该体系结构的稳定性由强到弱的排序为:VIa(2)>Ia(4)>IIa(4)>Ia(2)>IIa(2)>VIIa(2)>VIIa(4)>IIIa(4)>IVa(4)>IIIa(2)>Va(2)>IVa(2)。由以上分析可见具有C2V对称性的二重态的蝴蝶结构VIa为基态结构,键长F1-La1和La1-La2分别为0.231 3 nm和0.342 6 nm,键角La1-F1-La2为96°,双面角F1-La2-La1-F2为126°,能量为-1 071.184 047 9 a.u.。

图团簇的平均结合能
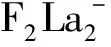
能隙的大小反映了电子发生轨道跃迁的能力,代表体系参与化学反应的强与弱。计算得到的两种体系稳定结构的最高占据轨道(HOMO)能级、最低空轨道(LUMO)能级以及两者之间的能隙(Egap)分别见表3和表4。团簇的能隙计算式为:Egap=ELUMO-EHOMO,其中Egap为团簇的能隙,ELUMO、EHOMO分别为最低空轨道和最高占据轨道的能量。表中也给出各种结构的振动频率,计算结果显示,所有稳定结构的频率均没有虚频,说明这些结构都是稳定存在的。由于中性团簇La2F2的价电子是偶数,其α轨道和β轨道的HOMO和LUMO的能量相等,表3未分别给出。由表3可见,三重态的结构I的能隙最大为2.051 eV,其化学活性较弱;单态结构VII的能隙最小为0.803 6 eV,其化学活性较强;三重态的基态(图1III)的能隙大小为1.475 eV。由于负一价团簇La2F2-1的价电子是奇数,其α轨道和β轨道的HOMO和LUMO的能量不相等,表4分别给出α轨道和β轨道的能量和能隙。研究化学活性的强弱,即是研究电子跃迁的难易程度,能隙越小跃迁越容易,在研究价电子数为奇数时的化学活性,要整体看每一种构型的α轨道和β轨道的能隙大小,小的能隙可用来作比较。由表4可见,四重态结构(图2 IIIa)的β轨道能隙较大为1.573 eV,其化学活性较弱;二重态结构VIIa的β轨道能隙较小为0.653 9 eV,其化学活性较强,容易与其它分子发生化学反应,可被用来制作多功能材料;基态(图2VIa)的α轨道和β轨道的能隙分别为1.685 eV和1.474 eV。

表3 La2F2团簇稳定结构的最高占有轨道能级HOMO,最低空轨道能级LUMO能量、能隙(Egap)及频率
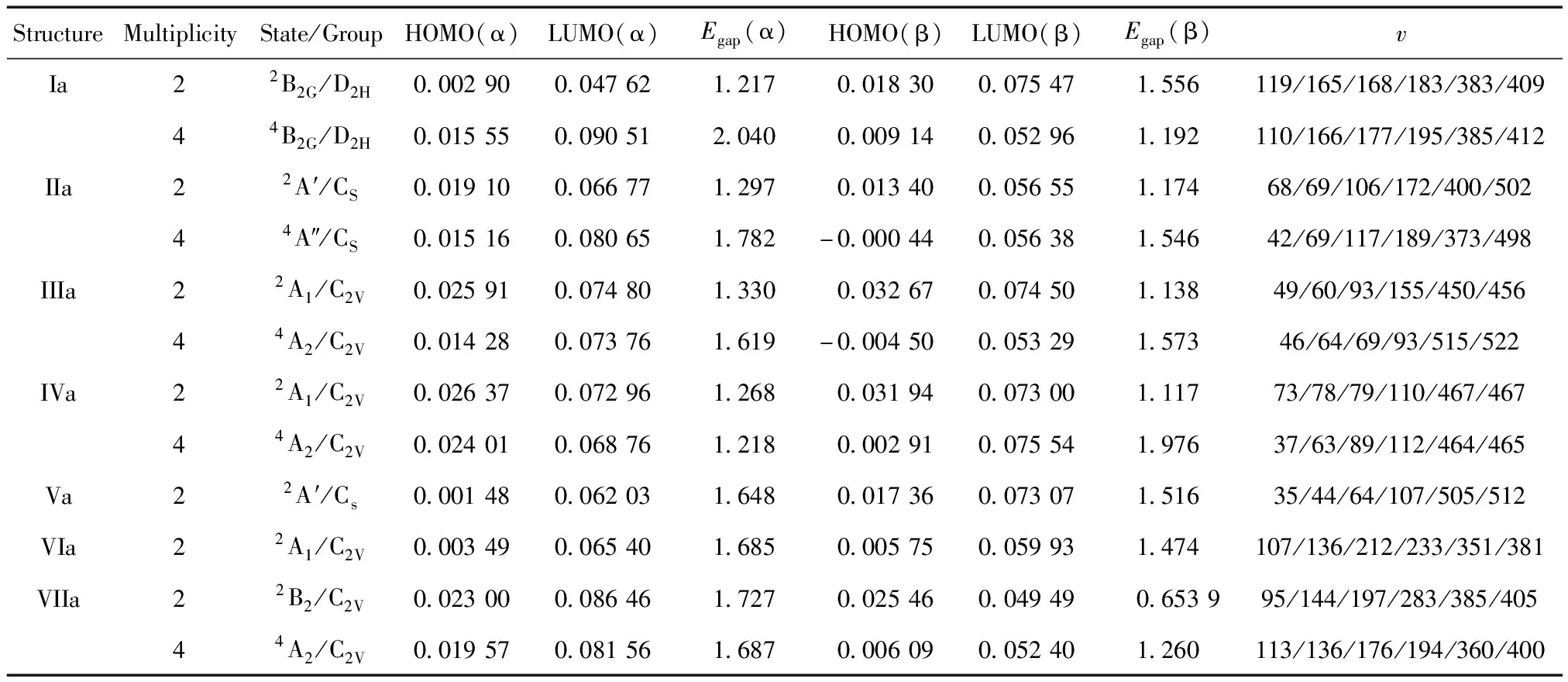
表4 La2F2-1 团簇稳定结构的最高占有轨道能级HOMO,最低空轨道能级LUMO能量、能隙(Egap)及频率
3 结 论
