ALK阳性非小细胞肺癌靶向治疗耐药机制及治疗管理的研究进展*
2022-04-06徐子钧胡继繁刘子玲
徐子钧 胡继繁 刘子玲
作者单位:吉林大学第一医院肿瘤中心肿瘤科(长春市130012)
肺癌因其高发病率及高死亡率被广泛关注。化疗为传统的治疗方法,随着基因检测及分子靶向治疗的发展,治疗模式也发生变化。目前,间变性淋巴瘤激酶(anaplastic lymphoma kinase,ALK)抑制剂为ALK突变阳性的晚期非小细胞肺癌(non-small cell lung carcinoma,NSCLC)患者的标准一线治疗方法,相关ALK 抑制剂已有三代药物的更新及发展,虽然其可提高无进展生存期(progression-free survival,PFS)和总生存(overall survival,OS)率,但治疗后的耐药机制及管理手段仍需进一步研究。
1 ALK 与NSCLC
1.1 NSCLC 流行病学及现状
现阶段肺癌仍是世界上最常见的恶性肿瘤,居癌症死亡原因的第1 位[1]。NSCLC 占所有肺癌病例的85%以上,其中约2/3 存在基因改变,1/2 患者可获得靶向治疗机会[2]。对治疗有显著反应的NSCLC 基因型包括表皮生长因子受体(epidermal growth factor receptor,EGFR)突变、ALK 突变、ROS 原癌基因1(ROS proto-oncogene 1,ROS1)重排等,本文旨在讨论ALK 突变相关内容。
1.2 ALK
1.2.1 ALK 生理表达及功能作用 虽然ALK 重排仅占NSCLC 的3%~7%[3],但大量的NSCLC 病例使得这一类患者全球每年新增约4 万例。ALK 可与多种配对基因融合重排,NSCLC 中最常见融合发生在棘皮动物微管相关类蛋白4-间变性淋巴瘤激酶(echinoderm microtubule-associated protein-like 4-anaplastic lymphoma kinase,EML4-ALK)。不同的ALK 融合蛋白的类型可以介导不同的信号输出,产生对ALK 抑制剂的不同敏感性及相关耐药性。
1.2.2 间变性淋巴瘤激酶酪氨酸激酶抑制剂 小分子酪氨酸激酶抑制剂(tyrosine kinase inhibitors,TKIs)阻断异常ALK 信号通路与受体酪氨酸激酶结合已被证实是抑制ALK阳性癌细胞生长的有效途径[4]。目前,crizotinib、ceritinib、alectinib、brigatinib 和lorlatinib 共5 种ALK 小分子抑制剂已被美国食品药品监督管理局(FDA)批准用于ALK阳性的NSCLC,ensartinib 作为首个国产间变性淋巴瘤激酶酪氨酸激酶抑制剂(anaplastic lymphoma kinase-tyrosine kinase inhibitors,ALK-TKIs)获中国药监局批准。
第一代ALK 抑制剂crizotinib,是ALK阳性NSCLC 分子靶向治疗的历史性里程碑,表明个体化方案制定在肿瘤治疗中至关重要。其优于传统化疗方案表现在明显的PFS、总缓解率(overall response rate,ORR)改善并且在不同治疗环境中均可看到优势[5],但缺乏血脑屏障穿透力。第二代ALK 抑制剂ceritinib、alectinib、brigatini、ensartinib、entrectinib 较一代有更多敏感突变作用点及更高ORR、PFS 及颅内活性,第三代ALK 抑制剂lorlatinib 可用于治疗几乎所有已知的ALK 抑制剂引起的耐药性突变。目前,三代ALK抑制剂作用靶点、耐药突变及颅内活性等情况,归纳见表1。然而类似于EGFR 的药物抗性,ALK 抑制剂治疗后的耐药性使得如何科学制定后续治疗策略成为亟待解决的问题。
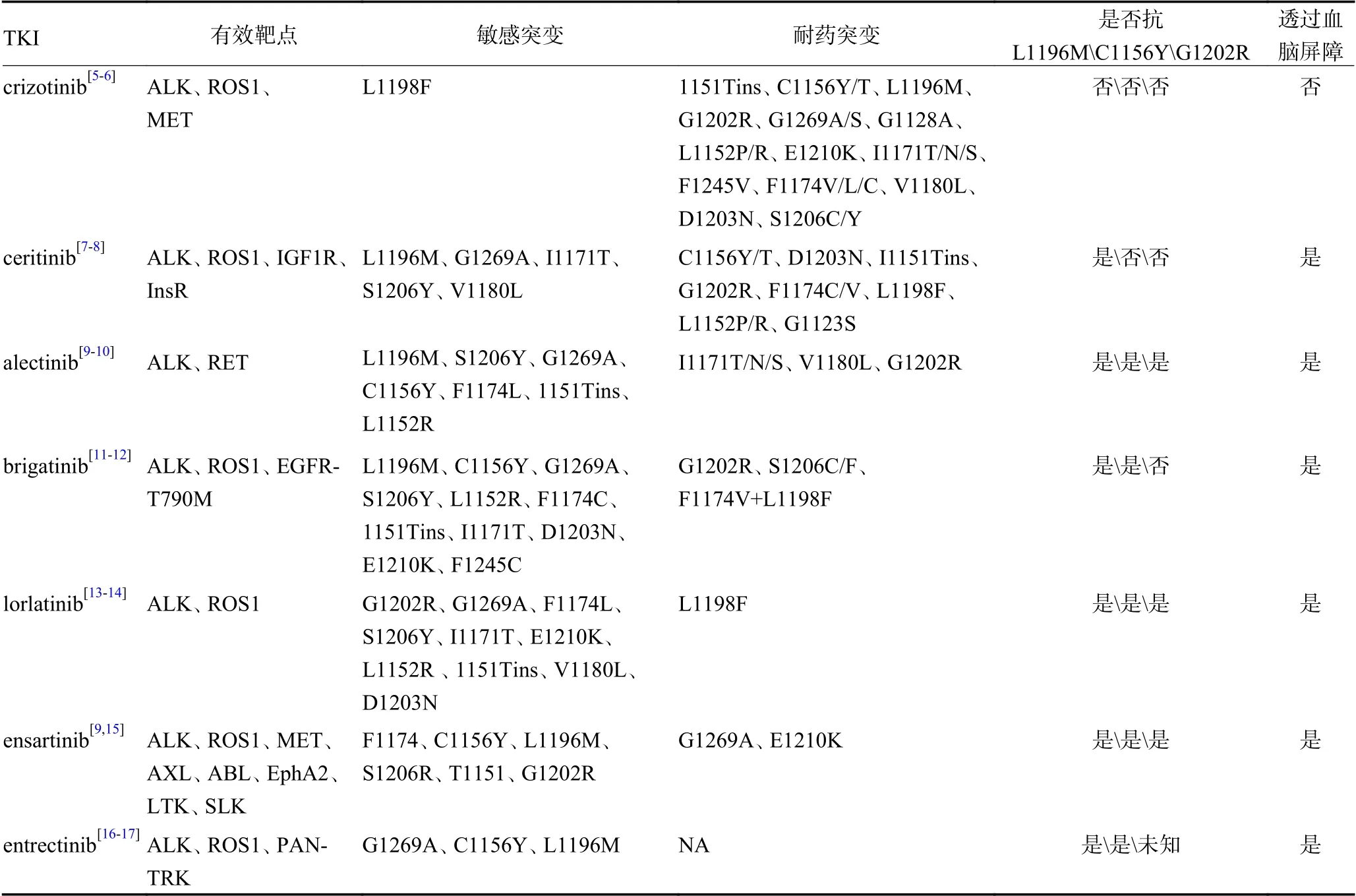
表1 ALK-TKIs 相关作用靶点及耐药突变
2 ALK-TKIs 耐药机制
多数患者在ALK 抑制剂使用1年后会出现耐药导致疾病进展。其中多为获得性耐药,患者起初对ALK 抑制剂有反应而后出现疾病进展,其机制分为ALK 依赖型及非依赖型耐药。
2.1 ALK 依赖型耐药
ALK 依赖型耐药最常见的是靶点激酶区二次突变[18],第一代crizotinib 中L1196M 突变,第二代ceritinib 中G1202R 突变,其位于ATP 结合口袋和药物结合位点附近直接阻碍与药物的结合[19]。而F1174V 突变[20]、C1156Y 突变[21]是通过改变ALK 蛋白的二级结构导致激酶结构域的构象改变从而降低药物亲和力产生耐药。序贯应用ALK 抑制剂可获得更好的疗效但复合突变的发生率也可能同时增加,有报道在接受crizotinib、ceritinib和alectinib连续治疗后检测到C1156Y 和I1171N 双重突变,以及在连续应用crizotinib 和brigatinib 治疗后检测E1210K 和D1203N存在突变[6]。
ALK 依赖型耐药另一种机制是ALK 基因的扩增。有研究[22]报道,使用crizotinib 的患者体内野生型EML4-ALK 基因扩增水平较高,但未发现二次突变。在间变性淋巴瘤中介导的耐药中也同样观察到ALK 基因拷贝数增加。但根据现有数据无法明确基因扩增导致获得性耐药的因素条件及临界触发值。
2.2 ALK 非依赖型耐药
ALK 非依赖型耐药主要指旁路信号通路激活,多种旁路机制可诱导耐药。有研究[23]报道,EGFR 激活作为crizotinib、alectinib 和ceritinib 的耐药机制。在alectinib 中观察到转化生长因子α 诱导表皮生长因子受体通路激活EGFR 信号转导通路,当其被敲除恢复了对alectinib 的敏感性。在小鼠移植瘤模型中,通过alectinib 及afatinib 双靶向ALK 及EGFR 观察到肿瘤体积缩小。Wilson 等[24]研究发现配体介导的HER2/3 激活和蛋白激酶C 激活是crizotinib 耐药的驱动因素。肝细胞生长因子诱导MET 活化刺激接头蛋白 GAB1 磷酸化并激活下游信号通路,是alectinib 旁路信号通路耐药的过程[25]。
非依赖型耐药机制还包括形态学的改变,如上皮间充质转化(epithelial mesenchymal transformation,EMT)以及向小细胞肺癌转化。在细胞实验及肿瘤样本中,分别证实了EMT 对第一代和第二代ALK 抑制剂均具有耐药性[6]。EMT 是一种独立的ALK 靶向耐药机制,目前暂无相关靶向治疗方法。
有研究[7]表明,在crizotinib 或ceritinib 耐药的NSCLC 中发现P-糖蛋白(P-gp)过表达,应用shRNA或药物下调或抑制P-gp 使耐药细胞重新对crizotinib 或ceritinib 敏感,同时P-gp 限制药物对中枢神经系统的渗透,这解释了为何crizotinib 或ceritinib 的颅内活性差,而alectinib 并非P-gp 的底物,其具有较强的颅内活性。
表观遗传学改变主要包括启动子DNA 高甲基化、全基因组DNA 低甲基化、组蛋白乙酰化和染色体结构异常。LncRNA 是由200 个核苷酸组成的一组具有低/非蛋白编码功能的RNA。lncRNA 异常表达参与转录和转录后调控等多种生物学功能并与耐药密切相关。肿瘤微环境(tumor microenvironment,TME)中肿瘤细胞内部变化与周围成分相互作用也可导致耐药。
3 耐药后治疗策略
对耐药机制的深入分析,将为开发更为有效的小分子抑制剂及设计化疗联合靶向治疗方案提供理论依据。克服耐药的策略主要集中在开发新一代的ALKTKI 上,并且可以对现有的治疗手段及药物进行合理的搭配组合,从而达到治疗的最佳效果。
3.1 ALK-TKI 的序贯应用
根据相关指南及推荐,第二代TKIs 为ALK阳性晚期NSCLC 的一线用药,虽然下一代的有效突变较前有所拓宽,但单药的应用仍无法规避耐药。不同的ALK 抑制剂具有不同的结构及作用靶点,需分析各自敏感及耐药突变,并在病程中重复活检。如患者一线应用第二代ALK-TKIs 复发复检耐药机制为ALK 依赖型则对第三代药物仍敏感,对于ALK 非依赖型耐药单纯序贯ALK 的抑制剂意义有限。联合其他治疗手段,如化疗或其他旁路通路第二激酶靶点抑制剂来治疗更具优势。另外,在重复活检中获得耐药突变谱后顺序或反序选择ALK 抑制剂,如检测到G1202R突变可选择的仅有lorlatinib,但有时需要反序选择,即使前期已耐药,如crizotinib 及ceritinib 耐药后序贯alectinib,但后者导致的I1171T 和V1180L 继发突变可通过ceritinib 来克服。提示未来使用两种不同ALK 抑制剂联合治疗的可能性。
但序贯治疗将增加复合突变概率,如1 例ALK阳性晚期NSCLC 患者先后应用crizotinib、ceritinib 及lorlatinib 后检测出存在C1156Y 和L1198F 双重突变,尽管lorlatinib 对C1156Y 突变敏感,但L1198F加入后产生的复合突变破坏了药物与激酶的结合导致耐药。体外研究证明[26],L1198F 突变对crizotinib 敏感,给予crizotinib 后患者获得了持久反应。针对该形式,提示更有效的长期策略是预先使用涵盖更多敏感突变的第三代ALK 抑制剂减少或防止靶向耐药,耐药后再行化疗、免疫治疗或临床试验。
3.2 ALK-TKI 联合其他治疗手段
3.2.1 联合旁路途径抑制物 临床研究[27]表明,在含有MEK 突变来源的ALK 重排NSCLC 细胞系使用MEK 抑制剂selumetinib 与ceritinib 联合可抑制肿瘤细胞增殖。另一项研究[28]证实,ALK/MEK 的双重阻断可克服ALK-TKIs 耐药并延缓耐药发生。从某种意义上说,ALK 抑制剂联合旁路途径抑制物可能会在将肺癌转变为慢性病方面提供机会。
3.2.2 联合抗血管生成药物 已有研究[29]证实,在ALK 改变的异种小鼠模型中VEGFR2 与分子靶向药物联合应用增强抗肿瘤作用,目前正处于临床试验阶段的ALK 抑制剂联合抗血管生成药物正在至少具有一处中枢神经系统病变的ALK阳性NSCLC 中进行。
3.2.3 联合免疫治疗 有研究[30]表明,EGFR-T790M、EML4-ALK、MET 过表达与程序性死亡配体-1(programmed death-ligand 1,PD-L1)表达增高显著相关,这为ALK-TKI 与免疫联合提供理论可能。有体外研究[31]表明,抗PD-L1 可能对ALK阳性NSCLC 具有潜在的治疗作用,但这一结论与部分病例报道不一致。截至目前,ALK-TKIs 与免疫疗法联合应用的疗效尚未明确。
3.2.4 联合局部治疗 有时患者仅为“寡进展”,一般指转移瘤数目≤5 个或受累器官≤2 个,是一段肿瘤生物侵袭性较温和的时期,是局限性原发灶与广泛转移之间的过渡阶段。这类患者更适用局部治疗方案如立体定向消融放射治疗(stereotactic ablation radiotherapy,SABR),其可以根除进展部位的耐药克隆,延缓耐药细胞生长,同时允许全身治疗继续足量应用。另外,无法耐受系统治疗、预备手术及脑转移者SABR可减小局部肿瘤体积进而加强对整体肿瘤的控制。
4 结语与展望
随着对肺癌耐药机制及相关分子生物学技术手段趋向精细灵敏,NSCLC 治疗格局正变得复杂性多样化,治疗方案的制定将是基因依赖性的。选择最佳的ALK-TKIs 治疗策略及耐药后联合方案,监测不良事件的后续管理,需要多学科联动的方法和专业知识积累。同时要认识到在NSCLC 的靶向治疗中肿瘤异质性是不断变化的,如癌症驱动基因的改变、表观遗传学改变等,重复活检综合分析肿瘤细胞及周围环境变化,动态监测肿瘤生长驱动因素等都显得尤为重要。
由于肺癌的高发病率及死亡率,易复发转移,有效的早期诊断及预后标志物具有重要意义。在表观遗传学研究中,通过鉴定转录因子基因启动子序列的DNA 高甲基化图谱,报告了同源框(HOX)相关基因,并有学者[32]提出HOX 是肺癌早期诊断和预后的生物标志物,间充质-HOX2 高表达与耐药性、预后和生存率有关。另外,有研究[33-35]在NSCLC 患者血清中检验出调控失调的lncRNA,如HotTip、BLACAT1 和SOX2/ANRIL,其可作为早期诊断标志物,表达水平与OS 相关,后续进一步对表观遗传学机制的探索将有望得到更多标志物,在提高NSCLC 的早期诊断率及预后方面作出新指引。
