治疗痛风中成药中掺杂西药成分吲哚美辛的含量测定
2022-04-02马德运
郑 静,马德运
(肇庆学院 食品与制药工程学院,广东 肇庆 526061)
痛风是一种与嘌呤代谢和尿酸直接相关的中老年疾病,常见于中年男性,且愈来愈趋向年轻化[1].痛风发作时,多数患者猝不及防,表现为突然半夜关节痛楚彻骨,且反复发作,疼痛难忍,浑身难受;少数患者有发热、寒颤、心慌和呕吐[2],甚至出现无法正常穿着等症状.此病为终身性疾病,目前不能完全根治.目前市场上销售的痛风西药主要有3大类:第一类是非甾体抗炎类,主要有双氯芬酸钠和保泰松片等,具有解热、镇痛和抗炎等作用[3];第二类是糖皮质激素类,主要有强的松、萘普生和地塞米松等,具有抗炎、抗毒和抗休克等作用[4];还有生物碱,主要是秋水仙碱,具有降温、消炎和抑制等作用[5].而中成药主要有:川芎嗪、新癀片、八珍丸、如意珍宝丸[6]、疏风定痛丸、六味地黄丸[7]、二十五味冰片散、复方罗布麻颗粒和痛风定胶囊等[8].
相对于中成药,西药的作用快,但其副作用亦不可小觑,特别是服用别嘌呤醇和秋水仙碱的患者,根据谢明[9]等人的调查,每100名痛风患者中就有33名患者产生不良反应,主要体现在消化道、肝脏、神经系统和泌尿系统等方面,均造成不同程度的伤害[10],所以病人更易于耐受和接受中成药,因此,抗痛风类中成药有很大需求量.从目前情况看,单纯使用中成药治病,效果较西药并不明显,因此为了提高疗效,治疗各类疾病的中成药制剂都有可能加入西药成分[11].然而中成药里西药成分的检测标准少之又少,加上我国对于这方面的监管、检测手段还不太完善,因此为此类药物提供检测评判标准显得尤为重要,研究这方面的检测标准对行业完善及群众健康有重大意义.
以市场上售卖的解热镇痛抗炎药、生物碱和糖皮质激素这3大类治疗痛风的西药为检测对象,选择5种化学药品,包括2种糖皮质激素类化学药品(醋酸泼尼松,醋酸地塞米松)、2种非甾体抗炎类化学药品(布洛芬,吲哚美辛)和1种生物碱(秋水仙碱),它们的作用都与治疗抗痛风中成药的作用相似,均可以达到解热镇痛抗炎的效果,可缓解病患的疼痛,且原料成本价格便宜低廉,能够大批量用于生产,是不少商家选择它们掺杂于中成药的重要原因之一[12].
薄层色谱法(Thin Layer Chromatography, TLC)系通过特定的薄层板,将待测溶液进行点样、展开和晾干等一系列操作进行分析,根据各比移值Rf作对比,用以快速进行药品的鉴别、分离和杂质检查的方法[13].高效液相色谱法(High Performance Liquid Chromatography, HPLC)系指通过高压输液泵将流动相泵入装有固定相的色谱柱中,由流动相将进样阀中的待测溶液带入柱内并进行分离,分离出的各成分逐一进入检测器进行检测,从而达到对试样的定性与定量的分析[14].本实验联合TLC和HPLC 2种方法,首先通过TLC快速对检测对象进行批量定性试验,进行初步筛选,节省大量时间,再使用HPLC对筛选出的对象进行精密定量测定,分析抗痛风药丸是否添加治疗痛风的西药及其含量[15],总结规律,为制定该类中成药是否掺杂西药成分的检测标准提供实验基础.
1 实验部分
1.1 药品、试剂与仪器
醋酸泼尼松、醋酸地塞米松、吲哚美辛、布洛芬等对照品均采购于中国食品药品研究所;秋水仙碱对照品采购于成都曼思特生物科技有限公司;抗痛风药丸购自市场.
有机试剂:氯仿、甲醇、乙酸乙酯、乙酸、氨水和无水乙醇均为分析纯;乙腈和甲醇为一级色谱纯,蒸馏水采购于市场.
主要仪器:电子天平(奥豪斯SE602F)、超声波清洗机(新芝SB-5200)、真空干燥箱(上海一恒DZF-6030)、三用紫外仪(安亭电子2F-2)、紫外可见光光度计(美谱达UV-1800),高效液相色谱仪(安捷伦Agilent 1200).
1.2 实验方法
1.2.1 薄层色谱法
1) 对照品溶液制备:称取醋酸地塞米松20.1 mg、秋水仙碱20.3 mg 和吲哚美辛20.0 mg,用移液管吸取10 mL 甲醇充分溶解,即得;同理,制备每mL 含20 mg 的布洛芬对照品溶液,称取醋酸泼尼松20.2 mg,加入10 mL氯仿溶解;即得对照品溶液.
2) 供试品溶液制备:将药丸于研钵研细,称取药品的一次服用量(2.169 8 g)于25 mL容量瓶中,加少量甲醇使溶解,于超声清洗机中超声提取30 min,蒸干,再用甲醇稀释定容至容量瓶刻度线.
3) 混合对照溶液制备:分别用移液管吸取2 mL上述对照品溶液和供试品溶液,置于同一小烧杯中,充分搅拌,摇匀,制成1:1(体积比)的混合溶液,即得.
4) 展开剂选择:醋酸地塞米松展开剂为三氯甲烷:乙酸乙酯:乙酸体积比30:10:1,醋酸泼尼松展开剂为饱和的乙酸乙酯,秋水仙碱、布洛芬和吲哚美辛的展开剂分别为氯仿:甲醇体积比19:1、乙酸乙酯:无水乙醇:氨水体积比50:16:1和氯仿:乙酸乙酯:冰醋酸体积比25:25:0.2.
5)实验测定:选择合适的溶液作为展开剂,选用同一块硅胶GF254薄层板,分别用毛细管吸取对照品溶液、供试品溶液和混合溶液各适量,点于板中,于展开剂中展开,取出,晾干;于254 nm紫外灯中检视.用尺子分别量取原点到斑点中心的距离和原点到溶剂前沿的距离,重复量取3次,详细记录,并计算出其比移值Rf(原点到斑点中心的距离与原点到溶剂前沿的距离的比值).
1.2.2 高效液相色谱法
1)对照品储备液制备:于10 mL容量瓶中,通过电子天平精密称取吲哚美辛0.012 8 g,加入少量甲醇溶解,摇匀,再用甲醇稀释定容至刻度线,摇匀,即得.
2)对照品溶液制备:用移液管精确量取1 mL上述对照品储备液,置于10 mL容量瓶中,加甲醇稀释至刻度线,摇匀,用0.22 μm微孔滤膜滤过,即得.
3)供试品溶液制备:将药丸于研钵研细,于100 mL 带塞锥形瓶中,使用电子天平精密称取样品2.193 7 g,精确加入50 mL甲醇,称定质量,于超声清洗机中超声提取30 min,冷却至室温,再次称重,缺失的量用甲醇补足,摇匀,用0.22 μm微孔滤膜滤过,取滤液作为供试品溶液.
4)色谱条件确定:色谱柱为Eclipse XDB-C18(4.6×250 mm,5 μm);流动相为甲醇:冰醋酸(0.1 mol/L)体积比为70:30;流速为0.6 mL/min;检测波长为320 nm;柱温为22℃;进样量为20 μL.
2 结果与讨论
2.1 薄层色谱法检测的结果
由图1 和表1 可见,5 种化学药品中,只有吲哚美辛对照品、供试品溶液及其两者的混合溶液(体积比1:1)在相同的位置呈现出相同颜色的斑点,其余均不同;初步认为抗痛风药丸中含有吲哚美辛.
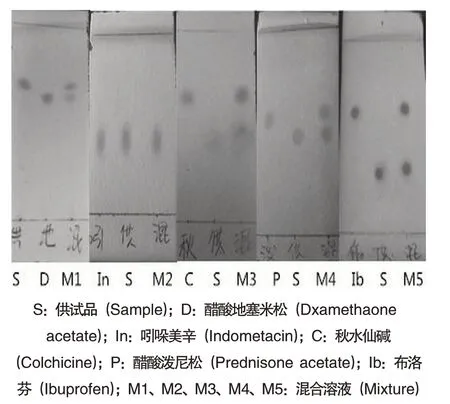
图1 薄层色谱图
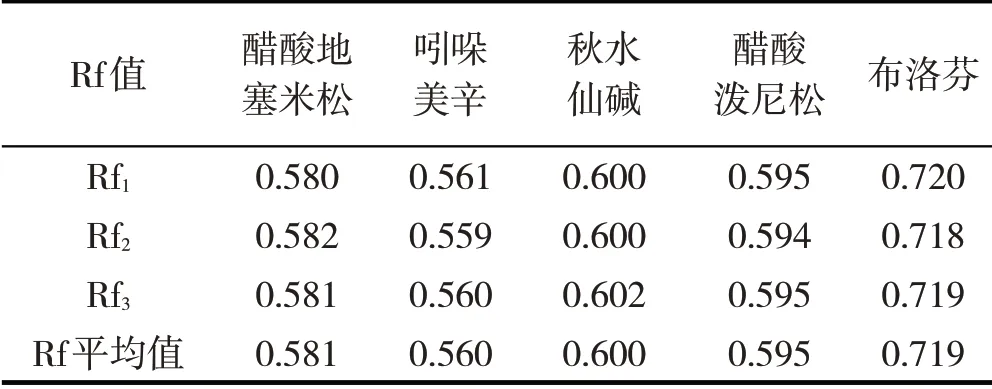
表1 各对照品Rf值
2.2 高效液相色谱法检测结果
2.2.1 专属性验证
通过手动进样针精密吸取对照品溶液、供试品溶液和空白对照溶液(甲醇)各30.0 µL,按照确定的色谱要求,手动进样20µL,得到谱图并比较,见图2,结果显示,吲哚美辛对照品和抗痛风药丸的保留时间分别为22.086 min 和22.145 min,且峰与峰之间分离度较好(R>1.5),无明显的重叠,理论塔板数(>2000)亦达到检测要求.
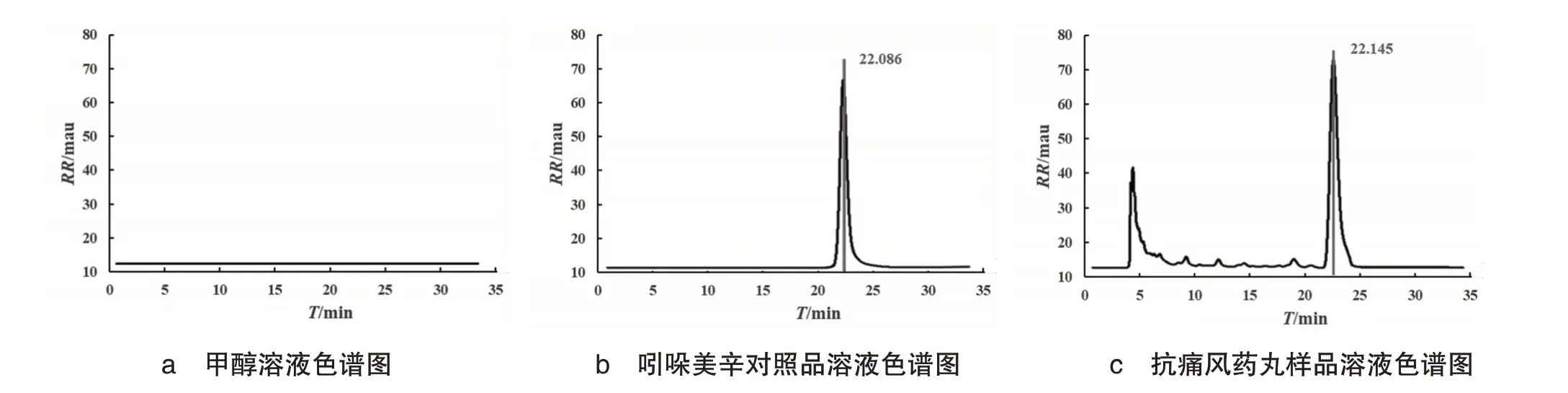
图2 溶液色谱图
2.2.2 线性实验
用3 个10 mL 容量瓶,使用移液管分别精确加入1 mL、1.5 mL、2 mL 上述的对照品储备液(1.28 mg/mL),用甲醇溶解并稀释至刻度,摇匀,制成浓度为0.13 mg/mL、0.19 mg/mL、0.26 mg/mL 的对照品溶液;同理,于3个25 mL容量瓶中,精确加入上述的对照品储备液1 mL、1.5 mL、2 mL,制成浓度为0.05 mg/mL、0.08 mg/mL、0.10 mg/mL的对照品溶液.
使用手动进样针精密吸取上述对照品溶液各30 μL,按照确定的色谱条件进针20 μL,详细记录每份对照品溶液的峰面积.
以浓度为横坐标,峰面积为纵坐标,绘制标准曲线,得吲哚美辛标准曲线回归方程y=22296x+410.75,证明吲哚美辛在浓度0.02~0.26 mg/mL 的范围内与其峰面积呈良好的线性关系,且相关系数R2=0.999 9.
结合上述实验可得每颗抗痛风药丸中约含6.4 mg吲哚美辛,即每克药丸中约含2.9 mg吲哚美辛.
2.2.3 精密度验证
精密吸取上述对照品溶液20µL,根据确定的色谱要求连续进针8针,测定并记录吲哚美辛对照品溶液的峰面积,得出相应的RSD值,见表2.

表2 吲哚美辛对照品精密度试验结果
结果显示,吲哚美辛的平均峰面积为3 543.95,RSD%为1.39,证明实验所用仪器精密度良好.
2.2.4 重复性验证
按上述操作步骤,选用相同批号的待测中成药配制样品工作液6 份,按照确定的色谱条件进样检测,记录每份样品溶液的峰面积,计算出相应的RSD值,结果见表3,证明实验方法较好.

表3 样品重复性试验结果
2.2.5 稳定性验证
按上述操作步骤,选用同一批号的待测中成药配制样品工作液,按要求进针,在时间分别为第0、1、2、3、4、5、6 h 测定峰面积,得出对应的RSD值,比较在0~6 h内样品溶液峰面积的偏差见表4.结果表明,抗痛风药丸在6 h内的含量测定较稳定.

表4 样品稳定性试验结果
2.2.6 对照品加样实验
将药丸于研钵研细,使用电子天平分别精密称取0.005 1 g、0.007 7 g 的供试品,置于100 mL 带塞锥形瓶中,精确加入甲醇50 mL,按上述操作步骤,制备供试品储备液A、B.
通过移液管各精密吸取5 mL上述供试品储备液A、B,置于10 mL 容量瓶中,加入已知吲哚美辛含量的样品溶液稀释至容量瓶刻度线,定容,摇匀,作为待测溶液A、B.
在确定的色谱条件下,分别精密吸取30µL上述待测溶液A、B和已知含量的供试品溶液,通过手动进样针手动进样20µL,每组重复进样3次,详细记录每组溶液的峰面积,计算出相应的回收率和RSD值,见表5.

表5 样品加样回收测定结果
结果表明,在相对浓度为80%、100%和120%3 个阶段时(对照品相对于供试品),平均回收率分别为97.60%、98.59%和100.80%,RSD%为0.03,证明对照品加样回收效果良好.
3 结论
试验为快速判定治疗痛风中成药中可能存在的化学药品成分,采取了薄层色谱法和高效液相色谱法结合的方式.基于薄层色谱结果,吲哚美辛对照品溶液和样品溶液及其两者的混合溶液在同一位置上出现相同的斑点,可初步确定该抗痛风药丸中含有吲哚美辛.最后通过高效液相色谱仪分析吲哚美辛的含量,结果显示所用方法线性良好(相关系数R2=0.999 9),具有较好的稳定性(RSD%为0.72)、重复性(RSD%为0.85)、精密度(RSD%为1.39)和回收率(RSD%为0.03).
