哌啶并咪唑啉二酮类化合物的除草活性研究
2022-04-02杨辉斌
刘 克,张 帆,杨辉斌,李 斌
(沈阳中化农药化工研发有限公司 新农药创制与开发国家重点实验室,辽宁沈阳 110021)
根据Phillips McDougall公司的数据统计,2019年全球作物用农药市场的销售额为598.27亿美元,其中除草剂销售额为261.75亿美元,占全球作物用农药市场的43.8%,是全球农药市场第一大产品类型[1]。
原卟啉原氧化酶(PPO)是叶绿素和血红素生物合成途径中的最后一种酶,能将原卟啉原IX催化氧化成原卟啉IX[2]。PPO抑制剂可抑制原卟啉原氧化酶的活性,导致原卟啉原IX不能氧化为原卟啉IX,大量积累的原卟啉原 IX流入到细胞质并在其中氧化为原卟啉IX,原卟啉IX在光和氧条件下产生单线态氧破坏细胞膜结构,最终造成植物死亡。PPO抑制剂具有高效、低毒、安全的作用特点[3],是新型除草剂研发中的重点和热点。20世纪60年代开发的二苯醚类除草剂是最早出现的 PPO抑制剂,1970-1980年期间,PPO抑制剂被大量研究[4],其结构类型主要包括二苯醚类和其他 PPO抑制剂两大类,其中1-杂环基-2,4,5-四取代苯类是其他PPO抑制剂中最主要的一类。自1969年第一个四取代苯类化合物噁草酮推广上市以来,目前已有20多个四取代苯类化合物进入产业化开发阶段,如丙炔氟草胺、氟噻乙草酯、三氟草嗪等。四取代苯类除草剂具有除草活性高、用量低、杀草谱广的优点,值得对其进行深入的研究与开发。
本课题组前期考察了不同的 1-位杂环基对四取代苯类化合物的除草活性的影响,其中 2-[2-氟-4-氯-5-炔丙氧基苯基]-四氢咪唑并[1,5-a]吡啶-1,3(2H, 5H)-二酮(a)、2-(2-氟-4-氯-5-炔丙氧基苯基)-四氢-[1,2,4]三唑并[1,2-a]哒嗪-1,3(2H)-二酮(b)、2-[2-氟-4-氯-5-炔丙氧基苯基]-4,5,6,7-四氢异吲哚-1,3(2H)-二酮(c)、1-(2-氟-4-氯-5-炔丙氧基苯基)-哌啶-2,6-二酮(d)和 3-(2-氟-4-氯-5-炔丙氧基苯基)-1-甲基-6-三氟甲基-2,4-(1H,3H)-嘧啶二酮(e)的除草活性较高[5](图1)。因为哌啶并咪唑啉二酮杂环易制得且原料成本低,所以本文选取化合物a为先导,对苯环5-位取代基部分进行变化,考察5-位取代物对苘麻生长的抑制活性,总结构效关系,为发现高除草活性的PPO抑制剂提供参考。结构通式如图2所示:
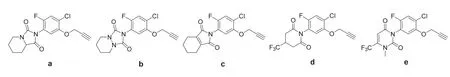
图1 高活性四取代苯类化合物
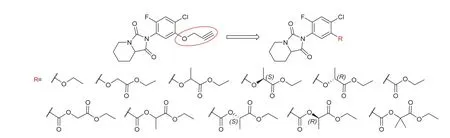
图2 哌啶并咪唑啉二酮类化合物的设计
化合物的合成路线如图3所示。
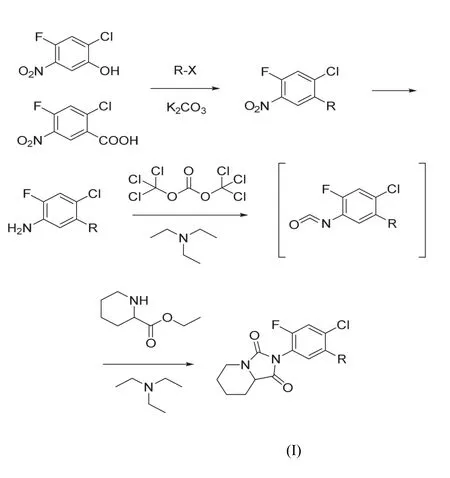
图3 哌啶并咪唑啉二酮类化合物的合成路线
以 2-氟-4-氯-5-硝基苯酚和 2-氟-4-氯-5-硝基苯甲酸为起始原料,经取代、硝基还原,制备异氰酸酯,环合得到目标化合物。
1 试验部分
1.1 仪器
Mercury 300核磁共振仪(TMS为内标,美国瓦里安公司);VG ZAB-HS质谱仪、旋光仪Autopol VI(美国鲁道夫公司);X4型显微熔点仪(北京第三光学仪器厂),温度计未经校正。
1.2 试剂
试剂均为市购分析纯。层析硅胶(GF245)薄板(涤纶片基,5×10 cm,浙江省台州市路桥四甲生化塑料厂);快速柱层析用硅胶(ZCX-II,粗孔,100~140目,青岛海洋化工厂)。
1.3 旋光测试
采用钠光谱的D线(589 nm)测定旋光度,测定管长度为1 dm,测定温度为20 ℃。称取1.0 g样品用丙酮配成浓度0.1 g/mL的供试溶液,将测定管用供试溶液冲洗数次,缓缓注入供试溶液,置于旋光计内检测读数,即得供试液的旋光度。用同法读取旋光度3次,取3次的平均数,按照下列公式计算,即得供试品的比旋度。
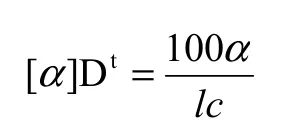
式中:[α]为比旋光度;D为钠光谱的D线;t为测定时的温度,℃;l为测定管长度,dm;a为测得的旋光度;c为溶液浓度,g/100 mL。
1.4 合成方法
1.4.1 2-(4-氯-5-乙氧基-2-氟苯基)四氢咪唑并[1,5-a]吡啶-1,3(2H,5H)-二酮(I-1)的制备
向反应瓶中加入 2-氯-4-氟-5-硝基苯酚(1.0 g,5.2 mmol)、DMF (20 mL)、碳酸钾(0.9 g, 6.2 mmol),滴加溴乙烷(0.6 g, 5.2 mmol),室温反应6 h,向反应液中加入乙酸乙酯(20 mL)和水(20 mL)萃取分液,有机相用饱和氯化钠溶液(10 mL)洗涤,无水硫酸镁干燥,减压蒸尽溶剂,得黄色固体,直接用于下一步。向反应瓶中加入 1-氯-2-乙氧基-5-氟-4-硝基苯(1.1 g, 5.0 mmol)、水合氯化亚锡(2.8 g, 12.5 mmol)、乙酸乙酯(20 mL),回流搅拌1 h,向反应液中加入氢氧化钠水溶液调节pH为7,过滤,滤液用乙酸乙酯和水萃取,有机相用饱和氯化钠溶液(10 mL)洗涤,无水硫酸镁干燥,减压蒸尽溶剂,得黑色固体,直接用于下一步。向反应瓶内加入 4-氯-5-乙氧基-2-氟苯胺(0.9 g, 5.0 mmol)、甲苯(20 mL)、三乙胺(0.8 g, 7.5 mmol),在冰浴冷却条件下缓慢滴加三光气(0.7 g, 2.5 mmol)的甲苯溶液,加热回流3.5 h,减压蒸尽溶剂,直接用于下一步。向反应瓶内加入2-哌啶甲酸乙酯(0.9 g, 5.5 mmol)、二氯甲烷(20 mL)、三乙胺(1.0 g, 10.0 mmol),滴加1-氯-2-乙氧基-5-氟-4-异氰酸酯苯的二氯甲烷溶液(5 mL)。室温搅拌1 h,向反应液中加入二氯甲烷(25 mL),分别用水(20 mL)、饱和碳酸氢钠溶液(20 mL)、饱和氯化钠溶液(20 mL)洗涤,有机相无水硫酸镁干燥,减压抽滤,减压蒸尽溶剂,残余物柱色谱提纯(淋洗液:乙酸乙酯/石油醚=1/3),得黄色黏液1.6 g,纯度98% (美国化学文摘登录号:85113-28-6[6]),收率95%。1H NMR (600 MHz, CDCl3)δ(ppm): 7.28 (d,J=9.0 Hz,1H), 6.83 (d,J=6.0 Hz, 1H), 4.27~4.23 (m, 1H), 4.07(q,J=7.2 Hz, 2H), 3.98~3.96 (m, 1H), 2.95~2.90 (m,1H), 2.33-2.29 (m, 1H), 2.09~2.06 (m, 1H), 1.82~1.79(m, 1H), 1.60~1.47 (m, 3H), 1.46 (t,J=7.2 Hz, 3H)。
按上述操作,分别制得化合物(I-2至I-5)。其物性及核磁数据如下:
2-(2-氯-5-(1,3-二氧六氢咪唑并[1,5-a]吡啶-2(3H)-基)-4-氟苯氧基)乙酸乙酯(I-2):黄色黏液(美国化学文摘登录号:101650-26-4[7]),收率80%;1H NMR (600 MHz, CDCl3)δ(ppm): 7.31 (d,J=9.0 Hz, 1H), 6.86 (d,J=6.6 Hz, 1H), 4.67 (s, 2H), 4.27 (q,J=7.2 Hz, 2H), 4.26~4.22 (m, 1H), 3.98~3.95 (m, 1H),2.96~2.89 (m, 1H), 2.32~2.29 (m, 1H), 2.09~2.04 (m,1H), 1.82~1.80 (m, 1H), 1.60~1.45 (m, 3H), 1.29 (t,J=7.2 Hz, 3H)。
2-(2-氯-5-(1,3-二氧六氢咪唑并[1,5-a]吡啶-2(3H)-基)-4-氟苯氧基)丙酸乙酯(I-3):黄色黏液,收率 78%;1H NMR (600 MHz, CDCl3)δ(ppm): 7.28(d,J=9.0 Hz, 1H), 6.87~6.85 (m, 1H), 4.72~4.68 (m,1H), 4.26~4.19 (m, 3H), 3.97~3.94 (m, 1H), 2.94~2.89(m, 1H), 2.31~2.29 (m, 1H), 2.09~2.04 (m, 1H),1.82~1.79 (m, 1H), 1.67 (d,J=6.6 Hz, 3H), 1.61~1.44(m, 3H), 1.26~1.23(m, 3H)。
(2S)-2-(2-氯-5-(1,3-二氧六氢咪唑并[1,5-a]吡啶-2(3H)-基)-4-氟苯氧基)丙酸乙酯(I-4):黄色黏液,[α]D20=-1.78 (c10, 丙酮),收率 91%;1H NMR(600 MHz, CDCl3)δ(ppm): 7.28 (d,J=9.0 Hz, 1H),6.88~6.86 (m, 1H), 4.72~4.68 (m, 1H), 4.26~4.19 (m,3H), 3.96~3.94 (m, 1H), 2.94~2.89 (m, 1H), 2.31~2.28(m, 1H), 2.09~2.04 (m, 1H), 1.82~1.79 (m, 1H), 1.67 (d,J=6.6 Hz, 3H), 1.61~1.44 (m, 3H), 1.26~1.23(m, 3H)。
(2R)-2-(2-氯-5-(1,3-二氧六氢咪唑并[1,5-a]吡啶-2(3H)-基)-4-氟苯氧基)丙酸乙酯(I-5):黄色黏液,[α]D20= +1.34 (c10, 丙酮),收率73%;1H NMR(600 MHz, CDCl3)δ(ppm): 7.28 (d,J=9.0 Hz, 1H),6.87~6.85 (m, 1H), 4.72~4.68 (m, 1H), 4.26~4.19 (m,3H), 3.96~3.94 (m, 1H), 2.94~2.89 (m, 1H), 2.31~2.28(m, 1H), 2.09~2.04 (m, 1H), 1.82~1.79 (m, 1H), 1.67 (d,J= 6.6 Hz, 3H), 1.60~1.44 (m, 3H), 1.26~1.23(m, 3H)。
1.4.2 2-氯-5-(1,3-二氧六氢咪唑并[1,5-a]吡啶-2(3H)-基)-4-氟苯甲酸乙酯(I-6)的合成
向反应瓶中加入2-氯-4-氟-5-硝基苯甲酸(1.1 g,5.2 mmol)、二氯甲烷(20 mL)、EDCI (2.0 g, 10.4 mmol)、DMAP (0.1 g, 1.0 mmol),滴加乙醇(0.2 g, 5.2 mmol),室温反应4 h,向反应液中加入二氯甲烷(20 mL),分别用水(20 mL)、饱和碳酸氢钠溶液(20 mL)和饱和氯化钠溶液(20 mL)洗涤,有机相无水硫酸镁干燥,减压蒸尽溶剂,得黄色固体,直接用于下一步。向反应瓶中加入 2-氯-4-氟-5-硝基苯甲酸乙酯(1.2 g,5.0 mmol)、铁粉(0.8 g, 15.0 mmol)、乙醇(20 mL)、四氢呋喃(10 mL),在冰浴冷却条件下缓慢滴加37%盐酸(3.0 g, 30.0 mmol)。室温搅拌2 h后减压蒸尽溶剂,加入乙酸乙酯(20 mL)和水(40 mL)萃取分液,有机相用饱和氯化钠溶液(10 mL)洗涤,无水硫酸镁干燥,减压蒸尽溶剂,得棕色固体,直接用于下一步。向反应瓶内加入2-氯-4-氟-5-氨基苯甲酸乙酯(1.1 g,5.0 mmol)、甲苯(20 mL)、三乙胺(0.8 g, 7.5 mmol),在冰浴冷却条件下缓慢滴加三光气(0.7 g, 2.5 mmol)的甲苯溶液,加热回流3.5 h,减压蒸尽溶剂,直接用于下一步。向反应瓶内加入2-哌啶甲酸乙酯(0.9 g,5.5 mmol)、二氯甲烷(20 mL)、三乙胺(1.0 g, 10.0 mmol),滴加2-氯-4-氟-5-异氰酸酯苯甲酸乙酯的二氯甲烷溶液(5 mL)。室温下搅拌1 h,向反应液中加入二氯甲烷(25 mL),分别用水(20 mL)、饱和碳酸氢钠溶液(20 mL)和饱和氯化钠溶液(20 mL)洗涤,有机相无水硫酸镁干燥,减压抽滤,减压蒸尽溶剂,残余物柱色谱提纯(淋洗液:乙酸乙酯/石油醚=1/3),得白色固体 1.5 g,纯度 98%,m.p.100~102 ℃(lit.102~103 ℃[8],美国化学文摘登录号:91623-94-8 ),收率 79%。1H NMR (600 MHz, CDCl3)δ(ppm): 7.91 (d,J=7.8 Hz, 1H), 7.36 (d,J=9.0 Hz, 1H), 4.38 (q,J=7.2 Hz, 2H), 4.27~4.24 (m, 1H), 4.00~3.98 (m, 1H),2.96~2.91 (m, 1H), 2.33~2.30 (m, 1H), 2.09~2.06 (m,1H), 1.83~1.80 (m, 1H), 1.61~1.47 (m, 3H), 1.39 (t,J=7.2 Hz, 3H)。
按上述操作,分别制得化合物(I-7至I-11)。其物性及核磁数据如下:
2-乙氧基-2-氧乙基 2-氯-5-(1,3-二氧六氢咪唑并[1,5-a]吡啶-2(3H)-基)-4-氟苯甲酸酯(I-7):黄色黏液,收率85%;1H NMR (600 MHz, CDCl3)δ(ppm): 8.05 (d,J=7.8 Hz, 1H), 7.39 (d,J= 9.0 Hz, 1H),4.83 (t,J=16.2 Hz, 2H), 4.26 (q,J=7.2 Hz, 2H),4.27~4.23 (m, 1H), 4.00~3.98 (m, 1H), 2.96~2.91 (m,1H), 2.33~2.30 (m, 1H), 2.09~2.06 (m, 1H), 1.83~1.80(m, 1H), 1.60~1.47 (m, 3H), 1.30 (t,J=7.2 Hz, 3H)。
1-乙氧基-1-氧丙烷-2-基 2-氯-5-(1,3-二氧六氢咪唑并[1,5-a]吡啶-2(3H)-基)-4-氟苯甲酸酯(I-8):黄色黏液,收率66%;1H NMR (600 MHz, CDCl3)δ(ppm): 8.00 (d,J=7.8 Hz, 1H), 7.37 (d,J=9.0 Hz, 1H),5.31 (q,J=7.2 Hz, 1H), 4.27~4.21 (m, 3H), 4.01~3.98(m, 1H), 2.96~2.91 (m, 1H), 2.33~2.30 (m, 1H),2.09~2.07 (m, 1H), 1.83~1.81 (m, 1H), 1.62(d,J=7.2 Hz,3H), 1.59~1.48 (m, 3H), 1.29 (t,J=7.2 Hz, 3H)。
(S)-1-乙氧基-1-氧代丙烷-2-基 2-氯-5-(1,3-二氧六氢咪唑并[1,5-a]吡啶-2(3H)-基)-4-氟苯甲酸酯(I-9):黄色黏液,[α]D20=-2.41(c10, 丙酮),收率70%;1H NMR (600 MHz, CDCl3)δ(ppm): 8.00 (d,J=7.8 Hz, 1H), 7.37 (d,J=9.0 Hz, 1H), 5.31 (q,J=7.2 Hz, 1H), 4.27~4.21 (m, 3H), 4.00~3.98 (m, 1H),2.96~2.91 (m, 1H), 2.33~2.30 (m, 1H), 2.09~2.07 (m,1H), 1.83~1.81 (m, 1H), 1.62(d,J=7.2 Hz, 3H),1.59~1.48 (m, 3H), 1.29 (t,J=7.2 Hz, 3H)。
(R)-1-乙氧基-1-氧代丙烷-2-基 2-氯-5-(1,3-二氧六氢咪唑并[1,5-a]吡啶-2(3H)-基)-4-氟苯甲酸酯(I-10):黄色黏液,[α]D20= +1.20 (c10, 丙酮),收率 68%;1H NMR (600 MHz, CDCl3)δ(ppm): 8.00 (d,J=7.8 Hz, 1H), 7.37 (d,J=9.0 Hz, 1H), 5.31 (q,J=7.2 Hz, 1H), 4.27~4.21 (m, 3H), 4.00~3.98 (m, 1H),2.96~2.91 (m, 1H), 2.33~2.30 (m, 1H), 2.09~2.07 (m,1H), 1.83~1.81 (m, 1H), 1.62 (d,J=7.2 Hz, 3H),1.58~1.47 (m, 3H), 1.29 (t,J= 7.2 Hz, 3H)。
1-乙氧基-2-甲基-1-氧丙烷-2-基 2-氯-5-(1,3-二氧六氢咪唑[1,5-a]吡啶-2(3H)-基)-4-氟苯甲酸酯(I-11):黄色黏液,收率 22%;1H NMR (600 MHz,CDCl3)δ(ppm): 7.89 (d,J=7.8 Hz, 1H), 7.36 (d,J=9.6 Hz, 1H), 4.27~4.20 (m, 3H), 4.01~3.98 (m, 1H),2.95~2.92 (m, 1H), 2.33~2.30 (m, 1H), 2.10~2.07 (m,1H), 1.83~1.81 (m, 1H), 1.69(s, 6H), 1.61~1.47 (m,3H), 1.26 (t,J=4.2 Hz, 3H)。
1.5 除草活性的测试
按设计剂量,分别称取1~8 mg供试样品于称量瓶中,加入1 mL丙酮使其充分溶解,然后加入含有1‰吐温80的静置自来水,搅拌均匀后得到所需浓度的样品溶液。以丙酮/水(1/3,含1‰吐温80)的处理作为空白对照。
将定量的苘麻种子分别播种于直径为 7 cm的装有营养土的纸杯中,播后覆土1 cm。种子发芽并生长 10~21 d,是处理前具有一系列生育阶段的试材。然后选择大小、生育阶段一致的试材,于 2~3叶期进行处理,处理后置于温室并浇水。试验用Airbrush喷雾机进行处理(喷液量100 mL/m2)。试验设3次重复。处理后置于温室按常规方法培养,定期观察苘麻的生长发育情况。于处理后15 d目测调查供试药剂对苘麻的抑制生长效果,0为无抑制,100%为完全抑制。结果见表1。
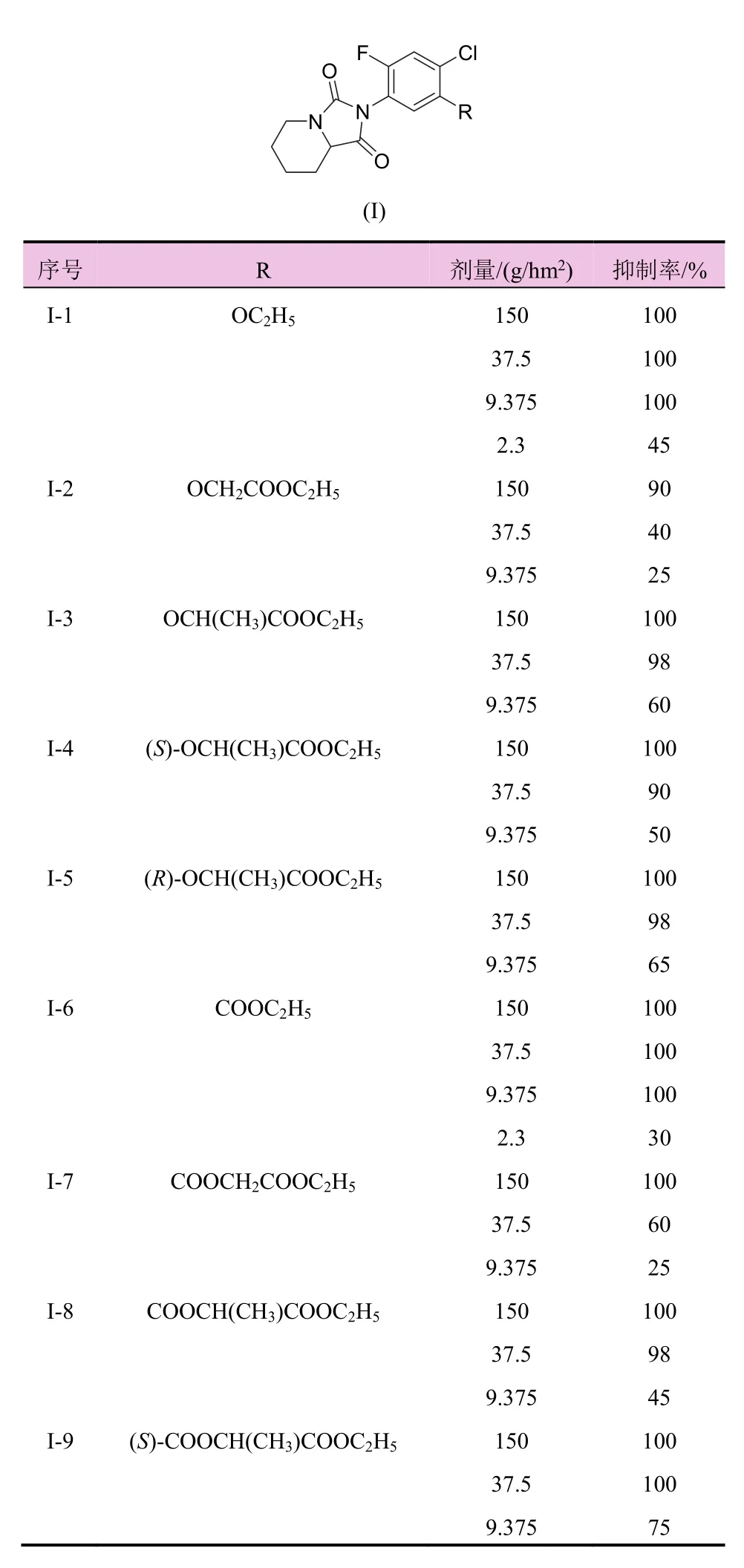
表1 哌啶并咪唑啉二酮类化合物的结构及除草活性
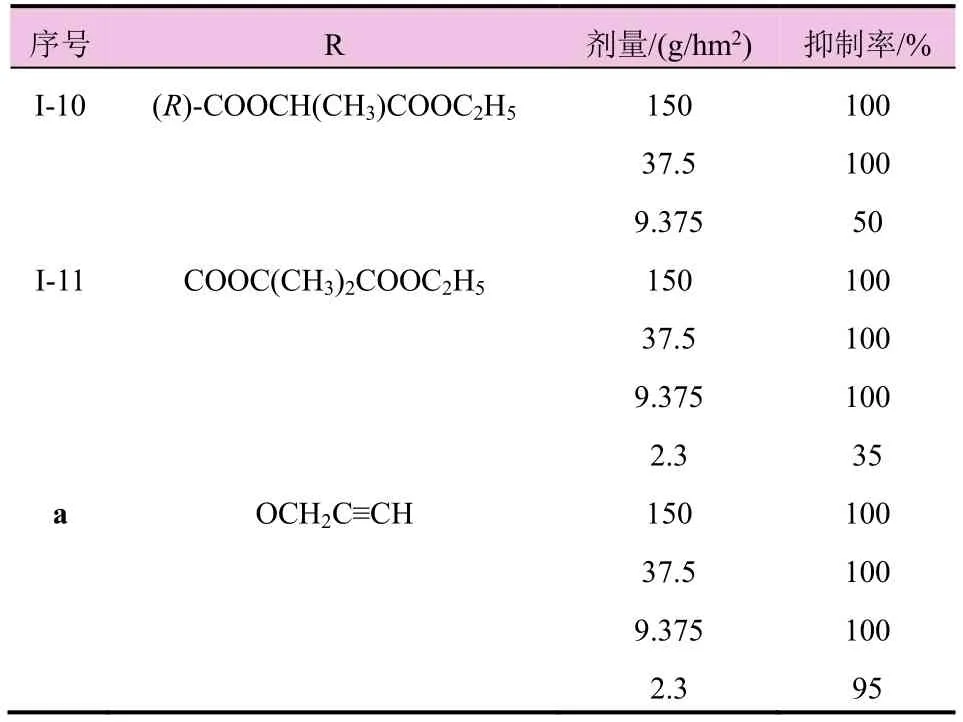
续表1
2 结果与讨论
根据表1中哌啶并咪唑啉二酮类化合物的结构及其对苘麻的抑制活性分析,得出如下构效关系:苯环5-位氧乙基化合物(化合物I-1、I-6)有较高的除草活性,在 9.375 g/hm2剂量下与先导化合物(a)相当,继续延长链(化合物I-2、I-7)后除草活性降低,说明增加侧链长度会使除草活性降低。
在侧链中引入甲基的支链化合物,其除草活性与甲基数量相关,当引入 1个甲基时(化合物 I-3、I-8),化合物表现出中等强度的除草活性,引入第2个甲基形成偕甲基化合物时(化合物I-11),化合物表现出较高的除草活性,在侧链中引入支链会增加除草活性,可能与占据靶点附近小的疏水口袋相关。
在侧链中引入手性碳后,对映异构体(化合物I-4 vs化合物I-5;化合物I-9 vs化合物I-10)之间的活性无显著差异。说明手性侧链对除草活性影响较小。
在2.3 g/hm2剂量下,化合物I-1、I-6、I-11比先导化合物a的除草活性均低,说明炔丙氧基对保持高除草活性有利。
3 结 论
本文设计合成了 11个哌啶并咪唑啉二酮类化合物,研究了苯环5-位取代基变化对除草活性的影响。当苯环5-位为乙氧基或乙氧羰基时,除草活性较好。化合物I-1和化合物I-6在9.375 g/hm2剂量下,对苘麻生长抑制率为 100%。当苯环 5-位为空间位阻较大的取代基时,除草活性较差。
本研究结果表明:当把苯环上的炔丙基变化为取代烷基后,除草活性降低。本文后续的研究重点将在以炔丙基和乙基为先导的基础上进行新化合物的设计与合成,以发现更高除草活性的化合物。
