2-氨基苯并咪唑衍生物的合成及抑菌活性初步研究
2022-04-02冯宇喆代文哲刘根炎巨修练
冯宇喆,代文哲,刘根炎,巨修练
(武汉工程大学 化工与制药学院,湖北武汉 430205)
农药是保障粮食丰产的有效手段之一,开发更加安全、高效、经济和使用方便的产品成为当今农药发展的主要方向。随着全球人口数量的不断增加,对粮食及果蔬的需求也随之增长,加之地球变暖,导致各种病原菌的危害愈来愈突出,因此,对杀菌剂的需求也不断增加。苯并咪唑类化合物由于其自身的优良特性,在材料、生物、医药、农药等领域均有广泛应用[1-3]。该类化合物在杀菌剂中占有重要地位,主要品种有多菌灵、丙硫多菌灵、苯菌灵、噻菌灵、麦穗宁等,具有广泛的杀菌活性,在防治农作物、水果、蔬菜、棉花等的病害防治方面发挥着重要作用[4-6]。
灰霉病病原菌(Botrytis cinerea)寄主范围广,具有繁殖速度快、产孢量大、遗传变异大和适合度高等特点,可以引起番茄、黄瓜、草莓等蔬果及花卉发生灰霉病,极大地影响了蔬菜瓜果的产量[7-9]。近年来,灰霉病以化学防治为主,目前普遍使用的药剂品种有多菌灵、苯菌灵、速克灵等。由于部分杀菌剂的广泛长期单一使用,导致病原菌对这些药剂产生了抗性,防治效果降低了,农用成本增加了[10-12],因此,开发新型对灰霉病病原菌具有较好防治效果的杀菌剂具有重要意义。
在总结文献的基础上,本研究采用了多菌灵和苯菌灵的公共骨架结构(2-氨基苯并咪唑)作为所设计化合物的骨架,在参考了敌菌灵结构中芳环取代的特点后,利用活性基团拼合等原理,设计并合成了2-N-取代苄基苯并咪唑类化合物(图1)。合成路线以2-氨基苯并咪唑为起始原料,与不同取代的芳醛反应生成中间体N-(1H-苯并[d]咪唑-2-基)-1-取代苯基甲亚胺,然后还原得到目标产物[13-17]。并初步测定了12个目标产物对灰霉病病原菌的抑制活性。
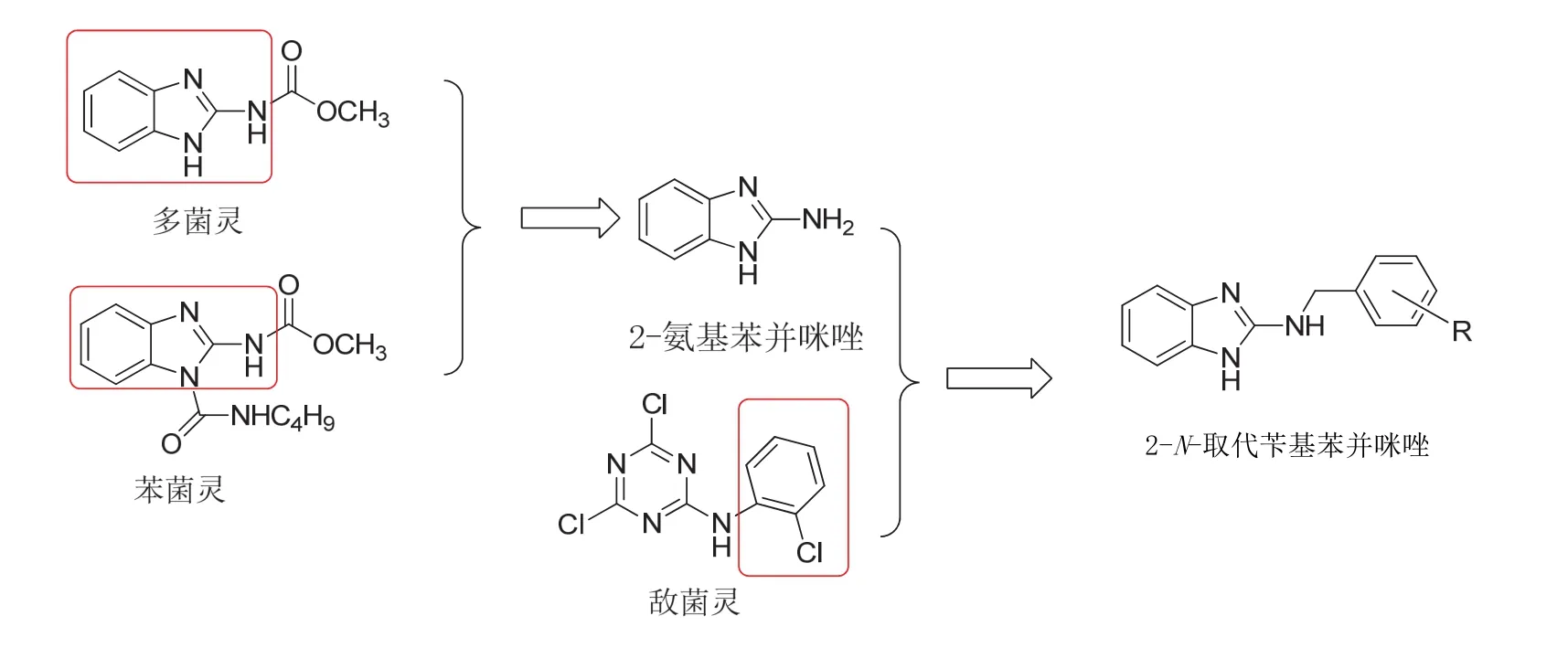
图1 目标化合物的设计
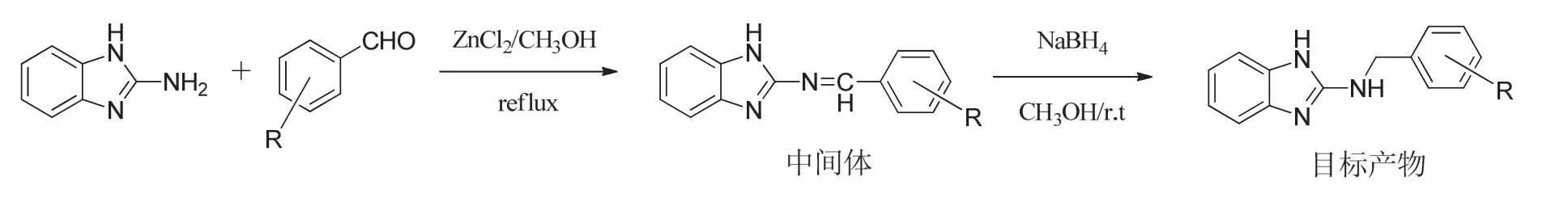
图2 目标化合物的合成路线
1 试验部分
1.1 仪器与试剂
RY-1型熔点仪(温度计未校正,天津天分分析仪器厂);Varian Mercury-VX 400型核磁共振仪(美国瓦里安公司);Thermo LTQ XL液质联用质谱仪(美国热电公司)。试验中所使用的主要试剂:2-氨基苯并咪唑、3-甲基苯甲醛、4-甲基苯甲醛、3-氟苯甲醛、4-氟苯甲醛、3-氯苯甲醛、4-氯苯甲醛、3-溴苯甲醛、4-溴苯甲醛、3,4-二氯苯甲醛、2,4-二氯苯甲醛、3,5-二氯苯甲醛、2,4-二氯苯甲醛(均为化学纯,国药集团),除特别注明外,均未经进一步处理直接使用。
1.2 合成方法
1.2.1 目标化合物N-(3-甲基苄基)-1H-苯并[d]咪唑-2-胺(1)的合成
将2-氨基苯并咪唑1.50 g(11.27 mmol)及3-甲基苯甲醛1.37 g (11.42 mmol)加入到100 mL的单口瓶中,然后加入氯化锌80 mg,量取45 mL无水甲醇加入到上述反应瓶中搅拌溶解。将反应液升温至回流,TLC检测跟踪反应进程,石油醚∶乙酸乙酯=1∶1 (体积比)为展开剂。在反应过程中,发现反应溶液由无色逐渐变为黄色,经TLC检测,有新点产生,反应11 h后,发现反应难以进行完全。停止加热,将反应溶液冷却至室温,旋转蒸发脱溶,得到黄色固体2.95 g。将上述黄色固体用6 mL乙酸乙酯溶解,加入适量硅胶拌匀,采用300~400目的硅胶进行柱层析,洗脱剂:石油醚∶乙酸乙酯=5∶1 (体积比),收集仅含有产物点的洗脱液,旋转蒸发脱溶,最后得到中间体N-(1H-苯并[d]咪唑-2-基)-1-取代苯基甲亚胺,黄色固体0.757 g,产率29%。称取上述中间体N-(1H-苯并[d]咪唑-2-基)-1-取代苯基甲亚胺600 mg (2.55 mmol)、硼氢化钠308 mg (7.65 mmol),量取甲醇30 mL一并加入到100 mL单口瓶中,常温搅拌。搅拌过程中不断有少量的气体放出,溶液由黄色逐渐变为无色。反应3 h后,TLC检测,展开剂为石油醚∶乙酸乙酯=1∶2 (体积比),反应完毕,此时溶液颜色为粉红色。旋转蒸发脱溶后,加入50 mL乙酸乙酯溶解,用30 mL饱和食盐水洗涤。然后将有机相用无水硫酸钠干燥,抽滤,滤液旋蒸除去溶剂,所得到的固体经柱层析纯化,洗脱剂:石油醚∶乙酸乙酯=5∶1 (体积比),得到淡黄色固体0.331 g。产率 55%,mp:139~143 ℃。1H NMR(DMSO, 400 MHz),δ:2.28 (s, 3H, CH3),4.48 (d, 2H,J=8.0 Hz, CH2),6.85~7.18 (m, 9H, ArH),10.80 (s, 1H,NH)。MS (ESI):238 (M+1)+。
1.2.2 目标化合物N-(4-甲基苄基)-1H-苯并[d]咪唑-2-胺(2)的合成
参照 1.2.1节目标化合物(1)的合成方法得到N-(4-甲基苄基)-1H-苯并[d]咪唑-2-胺(2),白色固体,产率88%,mp:175~178 ℃。1H NMR (DMSO,400 MHz),δ:2.26 (s, 3H, CH3),4.46 (d, 2H,J=8.0 Hz,CH2),6.84~7.27 (m, 9H, ArH),10.79 (s, 1H, NH)。MS (ESI):238 (M+1)+。
1.2.3 目标化合物N-(3-氟苄基)-1H-苯并[d]咪唑-2-胺(3)的合成
参照 1.2.1节目标化合物(1)的合成方法得到N-(3-氟苄基)-1H-苯并[d]咪唑-2-胺(3),淡黄色固体,产率83%。mp:134~136 ℃。1H NMR (DMSO,400 MHz),δ:4.54 (d, 2H,J=8.0 Hz, CH2),6.86~7.39(m, 9H, ArH),10.88 (s, 1H, NH)。MS (ESI):242(M+1)+。
1.2.4 目标化合物N-(4-氟苄基)-1H-苯并[d]咪唑-2-胺(4)的合成
参照1.2.1节目标化合物(1)的合成方法得到N-(4-氟苄基)-1H-苯并[d]咪唑-2-胺(4),淡黄色固体,产率76%,mp:166~169 ℃。1H NMR (DMSO, 400 MHz),δ:4.49 (d, 2H,J=8.0 Hz, CH2),6.85~7.43 (m, 9H, ArH),10.83 (s, 1H, NH)。MS (ESI):242 (M+1)+。
1.2.5 目标化合物N-(3-氯苄基)-1H-苯并[d]咪唑-2-胺(5)的合成
参照1.2.1节目标化合物(1)的合成方法得到N-(3-氯苄基)-1H-苯并[d]咪唑-2-胺(5),淡黄色固体,产率78%,mp:160~164 ℃。1H NMR (DMSO, 400 MHz),δ:4.52 (d, 2H,J=8.0 Hz, CH2),6.85~7.43 (m, 9H, ArH),10.89 (s, 1H, NH)。MS (ESI):258 (M+1)+。
1.2.6 目标化合物N-(4-氯苄基)-1H-苯并[d]咪唑-2-胺(6)的合成
参照 1.2.1节目标化合物(1)的合成方法得到N-(4-氯苄基)-1H-苯并[d]咪唑-2-胺(6),白色固体,产率78%,mp:169~174 ℃。1H NMR (DMSO, 400 MHz),δ:4.50 (d, 2H,J=4.0 Hz, CH2),6.79~7.41 (m, 9H,ArH),10.86 (s, 1H, NH)。MS (ESI):258 (M+1)+。
1.2.7 目标化合物N-(3-溴苄基)-1H-苯并[d]咪唑-2-胺(7)的合成
参照 1.2.1节目标化合物(1)的合成方法得到N-(3-溴苄基)-1H-苯并[d]咪唑-2-胺(7),白色固体,产率75%,mp:161~165 ℃。1H NMR (DMSO, 400 MHz),δ:4.52 (d, 2H,J=4.0 Hz , CH2),6.85~7.56 (m, 9H,ArH),10.88 (s, 1H, NH)。MS (ESI):303 (M+1)+。
1.2.8 目标化合物N-(4-溴苄基)-1H-苯并[d]咪唑-2-胺(8)的合成
参照 1.2.1节目标化合物(1)的合成方法得到N-(4-溴苄基)-1H-苯并[d]咪唑-2-胺(8),白色固体,产率52%,mp:187~191 ℃。1H NMR (DMSO,400 MHz),δ:4.48 (d, 2H,J=4.0 Hz,CH2),6.84~7.52 (m,9H, ArH),10.86 (s, 1H, NH)。MS (ESI):303 (M+1)+。
1.2.9 目标化合物N-(3,4-二氯苄基)-1H-苯并[d]咪唑-2-胺(9)的合成
参照 1.2.1节目标化合物(1)的合成方法得到N-(3,4-二氯苄基)-1H-苯并[d]咪唑-2-胺(9),白色固体,产率73%,mp:212~216 ℃。1H NMR (DMSO,400 MHz),δ:4.51 (d, 2H,J=8.0 Hz , CH2),6.85~7.61(m, 8H, ArH),10.89 (s, 1H, NH)。MS (ESI):293(M+1)+。
1.2.10 目标化合物N-(2,4-二氯苄基)-1H-苯并[d]咪唑-2-胺(10)的合成
参照 1.2.1节目标化合物(1)的合成方法得到N-(2,4-二氯苄基)-1H-苯并[d]咪唑-2-胺(10),白色固体,产率 64%,mp:193~197 ℃。1H NMR (DMSO,400 MHz),δ:4.56 (d, 2H,J=4.0 Hz , CH2),6.87~7.61 (m,8H, ArH),10.90 (s, 1H, NH)。MS (ESI):293 (M+1)+。
1.2.11 目标化合物N-(3,5-二氯苄基)-1H-苯并[d]咪唑-2-胺(11)的合成
参照 1.2.1节目标化合物(1)的合成方法得到N-(3,5-二氯苄基)-1H-苯并[d]咪唑-2-胺(11),白色固体,产率 60%,mp:156~160 ℃。1H NMR (DMSO,400 MHz),δ:4.53 (d, 2H,J=4.0 Hz, CH2),6.86~7.46(m, 8H, ArH),10.93 (s, 1H, NH)。MS (ESI):293(M+1)+。
1.2.12 目标化合物N-(2,4-二羟基苄基)-1H-苯并[d]咪唑-2-胺(12)的合成
参照 1.2.1节目标化合物(1)的合成方法得到N-(2,4-二羟基苄基)-1H-苯并[d]咪唑-2-胺(12),白色固体,产率 71%,mp:146~150 ℃。1H NMR(DMSO, 400 MHz),δ:4.49 (d, 2H,J=8.0 Hz, CH2),6.19~7.28 (8H, ArH),9.12 (s, 1H, OH),10.94 (s, 1H,NH),11.76 (s, 1H, OH)。MS (ESI):256 (M+1)+。
1.3 活性测定
1.3.1 培养基
马铃薯葡萄糖琼脂培养基(PDA):马铃薯20 g,琼脂20 g,葡萄糖20 g,灭菌蒸馏水1 000 mL。
1.3.2 试验菌种
灰霉病病原菌(Botrytis cinerea),南京农业大学提供。
1.3.3 目标化合物对病原菌的毒力测定
采用菌丝生长速率法[18]测定目标化合物对灰霉病病原菌菌丝的生长抑制作用。将每个目标化合物均用丙酮配制成0.5、5、50 μg/mL的待测溶液,测定其对灰霉病病原菌的抑制活性。在无菌操作台上,用移液枪取 1 mL配制好的样品溶液分别加入到培养皿中,用丙酮作空白对照,然后用无菌玻璃注射器注射9 mL融化好的培养基,使药液与培养基充分混合均匀,放置并冷却到室温。然后在培养皿中间位置接种病原菌,每个处理重复3次,最后将培养皿放入28 ℃的恒温培养箱中进行培养。培养3 d后,利用十字交叉法测出菌落直径,计算直径的平均值。最后根据菌落的平均直径计算抑菌率[19]。

按照以上方法测定所合成的 12个目标化合物的抑菌率。
2 结果与讨论
目标化合物1-l2对灰霉病病原菌的活性测试结果如表1所示。
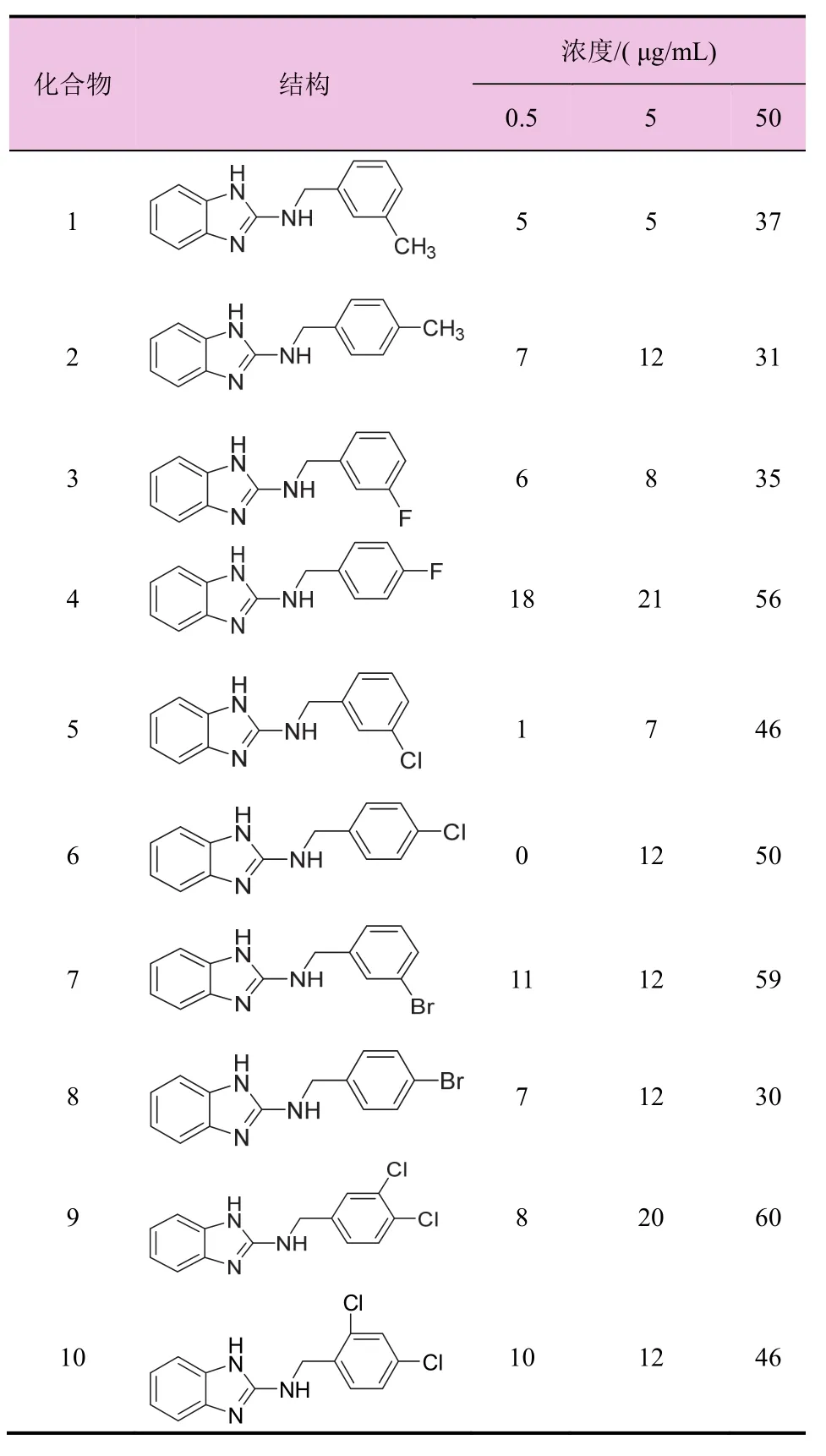
表1 目标化合物1-l2对灰霉病病原菌的抑菌率(%)
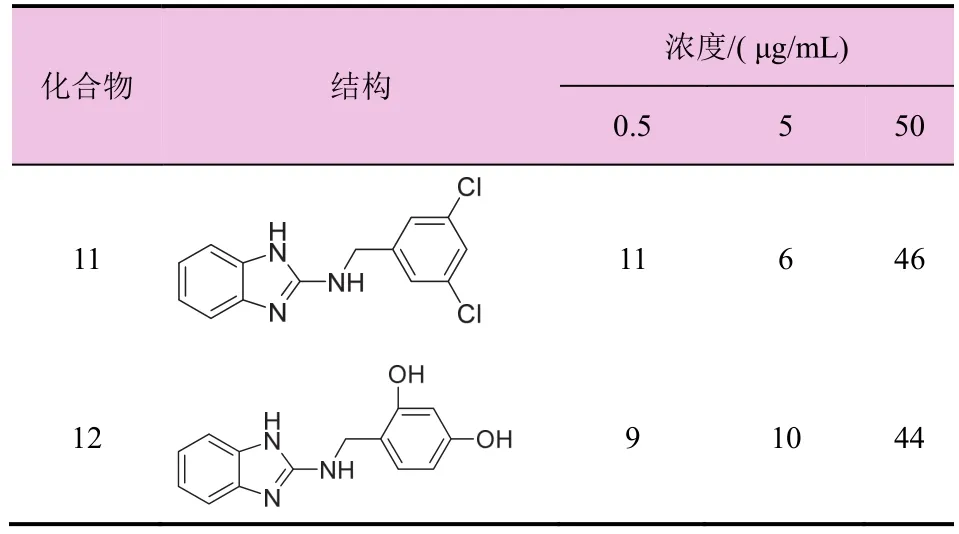
续表1
从表1可以看出,浓度在50 μg/mL时,所有化合物对灰霉病病原菌均有一定的抑制活性。2-氨基苯并咪唑衍生物中N取代苄基苯环对位溴取代的化合物8活性最低,抑菌率为30%;苯环甲基取代的化合物1和2活性相对较低,分别为37%和31%;苯环氯取代的化合物5和6的抑菌活性相近,分别为 46%和 50%;在苯环二氯取代化合物中,3,4-位二取代的化合物 9的抑菌活性分别高于 2,4-位和3,5-位取代化合物10和11的抑菌活性。化合物4、7、9的抑菌活性超过50%,化合物9活性最高达到60%,这些化合物可以进一步详细研究。
3 结 语
本研究针对2-氨基苯并咪唑的结构特点,利用活性结构拼合的原理,设计、合成了一类2-氨基苯并咪唑衍生物。以2-氨基苯并咪唑为起始原料,与不同取代的芳醛反应生成中间体N-(1H-苯并[d]咪唑-2-基)-1-取代苯基甲亚胺,然后用硼氢化钠还原得到12个目标产物。该合成方法原料易得,反应条件容易控制,所有合成的目标化合物经1H NMR及MS表征确认。通过菌落直径法初步研究了该类化合物对灰霉病病原菌的抑菌活性,结果表明所有目标化合物在50 μg/mL时对灰霉病病原菌具有一定的抑菌活性,化合物4、7、9的抑菌活性超过50%,化合物9活性最高达到60%。以上这些化合物可以进一步衍生化,进行结构与活性关系研究,希望开发出对灰霉病病原菌具有较高活性的新化合物。
