漆酶基因的克隆及其在产朊假丝酵母中的表达
2022-03-27高娃其布日萨初拉苏少锋吴青海白苏乐呼和郁彭
高娃,其布日,萨初拉,苏少锋,吴青海,白苏乐,呼和*,郁彭 *
(1.天津科技大学 生物工程学院,天津 300457;2.内蒙古自治区农牧业科学院 ,内蒙古 呼和浩特 010031)
农作物秸秆是宝贵的生物质资源,储存着大量的养分。充分利用秸秆资源,促进农业生态系统养分循环,是农业可持续发展的基础[1]。我国秸秆资源十分丰富,每年农作物秸秆的总量达6亿吨以上,但利用率非常低,大量秸秆无法被有效回收,只能就地焚烧,造成了环境污染和资源浪费,合理的加工处理可以有效提高农作物秸秆作为动物饲料原料的利用率[2-3]。造成秸秆饲料利用率低的主要原因是随着农作物的成熟,木质化程度增高,秸秆细胞壁中木质素含量也增加。木质素的结构复杂,木质素和半纤维素形成网状结构存在于植物细胞壁中,并将纤维素分子包埋在内,形成非常复杂的聚合物,使降解纤维素的酶与纤维素分子接触困难[4-6],很难在常规条件下降解。因此木质素被认为是限制秸秆有效利用的主要因素。
漆酶属多铜氧化酶家族[7],广泛存在于自然界中,是一种木质素降解酶。自然界中,漆酶主要来源于白腐真菌。白腐真菌是食用菌中的一类通过其特殊的酶系统高效降解木质素的微生物,将木质素降解为CO2和H2O[8],漆酶在此过程中起了重要作用。然而,自然界中真菌漆酶的产量低、酶活低、纯化难、生长周期长,限制了它的应用[9-10]。在蛋白质表达体系中,异源宿主高效表达更能满足工业化生产的需要。研究者们[11-14]通过漆酶的异源表达提高漆酶的产量,利用基因工程技术克隆不同来源的漆酶基因(lac),在不同宿主中异源表达,虽取得了一些成绩,但离理想的工业化生产还有一定的差距,有待于进一步提高酶蛋白的产量。
当前广泛使用的酵母表达系统有毕赤酵母和酿酒酵母,但各有缺陷。酿酒酵母大量表达时常常会发生质粒丢失现象;在发酵中通常有乙醇产生,很难进行高密度发酵。毕赤(甲醇)酵母虽能高表达、高分泌和高稳定[15-16],但不是食品酵母,诱导时需要加甲醇,无法被部分行业(食品和饲料)所接受[17];同时,甲醇耗氧多、产热大、有毒并且易燃,在工业生产中需进行防爆设计,导致生产成本增大。与之相比,产朊假丝酵母(Candida utilis,C.utilis)作为基因工程表达宿主具有很多优势,被食品药品监督管理局认证为食品级酵母,可运用于食品、饲料及制药业[18];C.utilis在严格好氧条件下不产生乙醇;C.utilis属多倍体,使用其基因组中重复序列(rDNA)作为整合载体的同源重组位点,可使外源基因与C.utilis染色体之间发生多拷贝同源重组[19]。因而C.utilis作为基因工程表达宿主具有很好的应用潜力。到目前,还未见白腐真菌来源的漆酶在C.utilis中异源表达的相关报道。
为此,本文从云芝栓孔菌中克隆lac基因,构建同源整合表达载体,以产朊假丝酵母(C.utilis)为宿主构建表达漆酶的基因工程产朊假丝酵母菌,为提高漆酶的产量和开发降解秸秆中木质素的微生物发酵剂提供基础资料。
1 材料和方法
1.1 菌株、试剂及培养基
云芝栓孔菌Trametes versicolor(BNCC 145690):北纳创联生物技术有限公司;pBR322质粒(3050):TaKaRa公司;产朊假丝酵母(31395):中国工业微生物菌种保藏管理中心;E.Z.N.ATMGel Extraction胶回收试剂盒:英国OMEGA公司;BcaBESTTMRNA PCR Kit、酵母总蛋白质提取试剂盒、PrimeSTAR GXL DNA Polymerase、载体 pMDTM19-T Vector:TaKaRa 公司;T4 DNA 连接酶、BamHI、SalI-HF、XbaI、NheI-HF、EcoRV、EcoRV-HF等内切酶类:Biolabs公司;Lyticase溶壁酶、TIANamp Yeast DNA kit酵母基因组DNA提取试剂盒、TIANprep Midi Plasmid质粒小提中量试剂盒:天根生化科技(北京)有限公司。
酵母浸出粉胨葡萄糖(yeast extract peptone dextrose,YPD)液体培养基:酵母提取物1%,蛋白胨2%,葡萄糖2%,加蒸馏水定容,121℃15min高压灭菌,备用。
YPD固体培养基:YPD液体培养基中添加2%的琼脂粉,灭菌备用。
产酶培养基(g/L):葡萄糖10,酒石酸铵0.1,KH2PO40.2,MgSO4·7H2O 0.5,MnSO40.035,CuSO4·5H2O 0.007,加水至 1 L,121℃灭菌 20 min。
50 mmol/L的琥珀酸缓冲液(pH4.5):0.81 g琥珀酸钠,定容至100 mL水中。称0.59 g琥珀酸,用配制的琥珀酸钠将pH值调至4.5。
0.4 mmol/L的愈创木酚:0.04 mmol(4.5 μL)愈创木酚,用1 mL 95%的乙醇溶解。
1.2 构建含lac基因的C.utilis表达载体
1.2.1 lac基因的克隆
提取云芝栓孔菌中总RNA,以提取的总RNA为模板进行逆转录-聚合酶链式反应(reverse transcription-polymerase chain reaction,RT-PCR)扩增,获得目的基因并与pMD19-T载体连接,转化提质粒,得到重组质粒。引物序列(5’-3’)如下。
Lac-R1:CAACATGTCGAGGTTTCACTCTC
Lac-R2:CCATTTACTGGTCGCTGGGGT
1.2.2 酵母三磷酸甘油醛脱氢酶基因启动子和终止子片段的克隆
根据GenBank数据库中C.utilis的GAP-p和GAP-t基因序列结合pBR322质粒序列特征设计引物,以C.utilis基因组为模板,经聚合酶链式反应(polymerase chain reaction,PCR)扩增获得酵母三磷酸甘油醛脱氢酶基因启动子(glyceraldehyde-3-phosphate dehydrogenase gene-prompter,GAP-p) 片段和终止子(glyceraldehyde-3-phosphate dehydrogenase gene-terminator,GAP-t)片段,将目的片段 GAP-p、GAP-t分别连接至载体pMD19-T simple vector,得质粒载体pTGP、pT-GT,转化大肠杆菌,阳性克隆进行鉴定并保存备用。引物序列(5’-3’)如下。
启动子:GAP-p1:GGATATCTTACAGCGAGCACTCAA
GAP-p2:GCTCTAGAATGTTGTTTGT
终止子:GAP-t1:CTAGCTAGCTATGACTTTTAT
GAP-t2:GGGATCCTTCATTCATCCCTCACTATCG
1.2.3 放线菌酮(cycloheximide,CYH)抗性基因的克隆
根据GenBank数据库中C.utilis的CYH敏感基因(L41)序列结合pBR322质粒序列特征设计一对引物P4/RM4,和1对反向互补引物P5/R5;以C.utilis基因组为模板,PCR扩增出L41上游片段和下游片段(两片段的一端具有碱基重叠区);以此上游片段、下游片段等量混合为模板,进行套叠PCR,即获得CYH抗性基因;将目的片段连接至载体pMD19-T simple vector上得到质粒载体pT-CYH,转化大肠杆菌,阳性克隆鉴定并保存备用。引物序列(5’-3’)如下。
L41:P4:CGTCGACAGTAAGTATGAAAAGAGC
RM4:GGGATCCGG GTTTGGTCTATGTTGCT
mL41:P5:AACCAAGCAAGTTTTCCAC
R5:GTGGAAAACTTGCTTGGTT
1.2.4 18S rDNA基因片段的克隆
根据GenBank数据库中C.utilis的18S rDNA基因序列结合pBR322质粒序列特征设计引物P3/R3,以C.utilis基因组为模板,经PCR扩增获得18S rDNA片段,将目的片段连接至载体pMD19-Tsimple vector上获得质粒载体pT-rD,转化大肠杆菌,阳性克隆进行鉴定并保存备用。引物序列(5’-3’)如下(18SrDNA基因片段)。
P3:CGATATCTGCCAGTAGTCATATGC
R3:CGATATCTGACTTGCGCTTACTAG
1.2.5 重组表达载体的构建
提取pT-GP、pT-GT和pBR322质粒,分别用EcoRV/XbaI,XbaI/BamHI和 EcoRV/BamHI酶进行酶切,回收目的片段,用T4 DNA连接酶将这3个片段同时连接,获得质粒pBR-GAP;提取pT-CYH和pBRGAP质粒,分别用BamHI/SalI酶酶切,回收目的片段,用T4 DNA连接酶进行连接,获得质粒pBR-G-L;提取pBR-G-L质粒和lac基因重组质粒,分别用XbaI/NheI酶酶切,回收目的片段,用T4 DNA连接酶进行连接,获得质粒pGQL;提取pT-rD和pGQL质粒,分别采用EcoRV-HF酶酶切,回收目的片段,用T4 DNA连接酶进行连接,获得含lac基因的重组表达载体pGQLR。
1.3 删除来自原核微生物的DNA序列
以pGQLR载体为模板进行引物设计,将片段序列分为两个片段,通过重叠延伸PCR试验技术[20]删除来自原核的DNA序列(包括抗药性标记Amp在内的细菌质粒序列的DNA片段)。重叠延伸PCR试验引物序列(5’-3’)如下。
P1-1S:CATGGTGGCAACGGGTAACGGGG
P1-1AS:GCATATGACTACTGGCAAGTAAGTATGAAAAGAGCCAAT
P2-1S:ATTGGCTCTTTTCATACTTACTTGCCAGTAGTCATATGC
P2-1AS:TGGTAGGCCACTATCCTACCATCGACAGTTGATAG
1.4 电转化C.utilis和转化子PCR鉴定
将已删除来自原核的DNA序列的整合表达载体电转化至C.utilis,构建工程菌株,命名为ZHQX1。
电转化操作在Raymond等[21]的方法基础上做了一些改良。酵母转化子提基因组,采用引物Lac-F1+1/Lac-R1+1进行PCR扩增,鉴定lac基因是否整合到C.utilis染色体DNA上。引物序列(5’-3’)如下。
Lac-F1+1:GCTCTAGAATGTCGAGGTTTCACTCTCTT
Lac-R1+1:CTAGCTAGCTTACTGGTCGCTGGGG
1.5 聚丙烯酰胺凝胶电泳(sodium dodecyl sulfatepolyacrylamide gel electrophoresis,SDS-PAGE)验证
1.6 转化子的稳定性试验
将工程菌ZHQX1用未添加CYH的YPD液体培养基进行接种传代培养后,选取培养第1天和第15天的菌液提取酵母基因组,采用PCR检测目的基因,测定外源基因遗传稳定性。
每次转接酵母的繁殖世代计算公式[22]为(lgN-lg0.1)/lg2。
1.7 漆酶的表达检测
分别取150 μL原始菌-云芝栓孔菌和工程菌ZHQX1于10 mL产酶培养基中,28℃、120 r/min振荡培养至对数期。分别在 24、48、72、96、192 h 时取菌液,7 200 r/min离心10 min,取上清液进行酶活测定。
漆酶的酶活测定:采用愈创木酚法。
缓冲溶液常用琥珀酸钠溶液(pH4.5),终浓度为50 mmol/L。底物愈创木酚的终浓度0.4 mmol/L,反应温度30℃,反应时间30 min,测定465 nm处OD值变化。
以100℃煮2 min~5 min的酶液为对照进行试验。酶活计算公式如下。

式中:V 为反应体积,mL;t为反应时间,min;Va为酶液体积,mL;ε为消光系数,9.3×103mol/(L·cm)。
2 结果与分析
2.1 lac基因的克隆
以提取自云芝栓孔菌总RNA为模板,采用BcaBESTTMRNA PCR Kit Ver.1.1进行RT-PCR扩增。PCR产物进行琼脂糖电泳检测,结果见图1。

图1 RT-PCR扩增结果Fig.1 Results of RT-PCR product
如图1所示,在1 000 bp~2 000 bp间扩增出的条带大小符合lac基因的预期值(约为1 500 bp)。
收集实验组新辅助放化疗后患者的治疗效果、临床分期变化情况及不良反应情况;收集并分析两组患者的手术切除率、保肛率、术后并发症及预后(生活质量及生存情况)。

图4 CYH基因的克隆结果Fig.4 The PCR result of CYH gene fragment
2.2 酵母三磷酸甘油醛脱氢酶启动子和终止子片段、放线菌酮(CYH)抗性基因和18S rDNA基因片段的克隆
以C.utilis基因组为模板,用相应引物进行PCR扩增,获得酵母GAP启动子(GAP-p)片段、酵母GAP终止子(GAP-t)片段、放线菌酮(CYH)抗性基因和18S rDNA基因片段,结果如图2~图5所示。

图2 GAP-p序列的克隆结果Fig.2 The PCR result of GAP-p fragment
从图2~图5可见,扩增出的条带大小与目的片段大小一致。
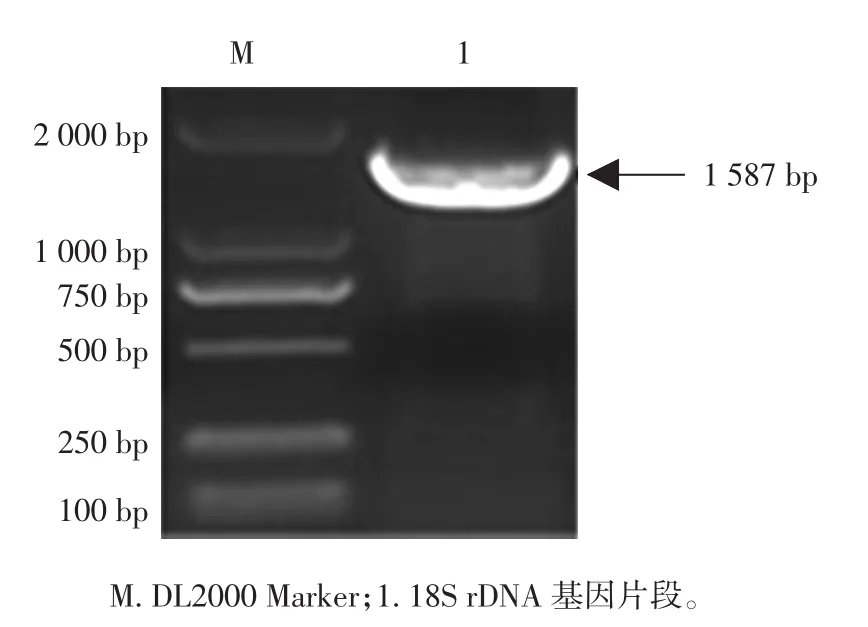
图5 18S rDNA基因片段的克隆结果Fig.5 The PCR result of 18S rDNA fragment
2.3 同源整合载体pGQLR的构建
整合载体pGQLR以pBR322为骨架载体,采用酶切连接法从3′端到5′端依次连接18S rDNA基因片段、酵母三磷酸甘油醛脱氢酶基因启动子序列GAP-p、lac基因、酵母三磷酸甘油醛脱氢酶基因终止子序列GAP-t和放线菌酮(CYH)抗性基因。提质粒后,采取酶切和PCR方法进行验证。整合载体pGQLR的构建结果见图6。

图3 GAP-t序列的克隆结果Fig.3 The PCR result of GAP-t fragment

图6 载体pGQLR的构建Fig.6 Consruction of the vector pGQLR
此同源整合表达载体命名为pGQLR。
2.4 转化子的PCR鉴定
胶回收重叠延伸PCR试验产物,将线性化的质粒在1.5 kV、25 uF、200 Ω条件下进行电转化至C.utilis。转化液离心浓缩后,全部涂在YPD固体平板(YPD+PTT+CYH)上,28℃、避光培养2 d~3 d后开始统计和挑取转化子。提取转化子的基因组,以其为模板,用特性引物Lac-F1+1和Lac-R1+1进行PCR扩增,结果见图7。
从图7可见,挑取的6个克隆扩增出的条带均与lac基因大小一致,说明lac基因成功整合到C.utilis染色体DNA上。
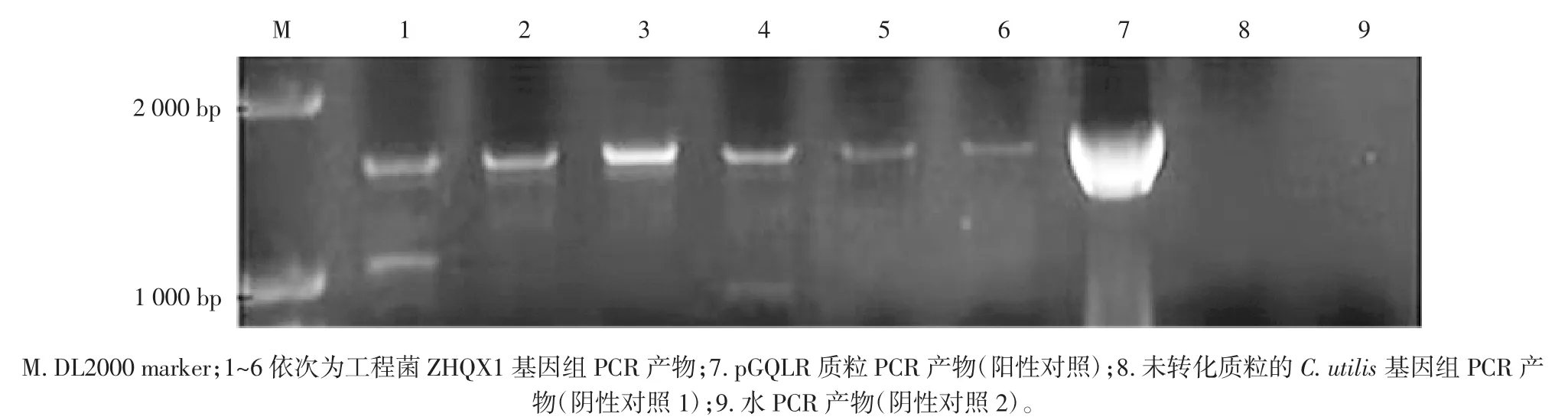
图7 转化子ZHQX1的PCR鉴定Fig.7 PCR Identification of ZHQX1
2.5 SDS-PAGE验证
经PCR鉴定为阳性的克隆进行培养,采用Yeast Protein Extraction Reagent试剂盒提取蛋白质,SDSPAGE法进一步鉴定,结果见图8。

图8 SDS-PAGE法检测ZHQX1蛋白的结果Fig.8 SDS-PAGE analysis of proteins secreted by ZHQX1
由图8可见,在55 kDa~70 kDa之间有明显的条带,与lac基因的蛋白分子量一致。说明lac基因已经整合到C.utilis的染色体DNA上。
2.6 转化子的稳定性试验
提取工程菌ZHQX1的基因组DNA,通过目的基因引物进行PCR扩增,琼脂糖凝胶电泳检测,结果如图9所示。
由图9可见,在连续传代100世代时仍含有目的条带,表明外源基因没有丢失,目的基因稳定存在,在传代培养过程中保持了较高的遗传稳定性。
2.7 漆酶的表达检测
不同培养时期云芝栓孔菌和ZHQX1的菌液,取上清做酶活测定,结果见表1。

表1 工程菌ZHQX1的酶活测定结果Table 1 Determination of enzyme activity of recombinant ZHQX1 U/L
从表1可看出,选取5个时间段取样测定工程菌漆酶酶活。在培养的前2 d原始菌的酶活为0。随着培养时间的延长,工程菌的漆酶酶活逐步升高,酶活在第96小时达到最高值1 078.9 U/L,是同时期原始菌—云芝栓孔菌酶活的4.8倍,在72 h的酶活是原始菌酶活的10倍。
3 结论与展望
本研究从白腐真菌—云芝栓孔菌中克隆了lac基因,用酶切连接法在pBR322载体的基础上从3′端到5′端依次连接18S rDNA基因片段、酵母三磷酸甘油醛脱氢酶启动子序列GAP-p、lac基因、酵母三磷酸甘油醛脱氢酶终止子序列GAP-t和放线菌酮(CYH)抗性基因,构建了含有lac基因的同源整合表达载体pGQLR。该载体的全部遗传元件均来自食品级酵母(C.utilis)和无毒无害的真菌(云芝栓孔菌),不携带抗生素抗性基因,不存在抗性基因的漂移、扩散及整合,也不携带其他有毒蛋白基因,不会对环境或人和动物带来生物安全性的危害。采用重叠延伸PCR试验技术删除同源整合表达载体中来自原核的DNA序列后电转化至C.utilis中,构建了表达漆酶的食品级基因工程菌ZHQX1。
ZHQX1在连续传代100世代时仍含有目的条带,表明外源基因没有丢失,在传代培养过程中保持了较高的遗传稳定性;培养96 h时,ZHQX1的漆酶酶活是同时期原始菌-云芝栓孔菌的漆酶酶活的4.8倍,达到1 078.9 U/L。
已知白腐真菌分泌几种与木质素降解相关的酶,即木质素过氧化物酶、漆酶、锰过氧化物酶、多功能过氧化物酶和多种辅助酶。多数分泌1种~2种降解木质素的酶,也有少数白腐菌可分泌3种降解木质素的酶[23],表明木质素的降解过程中主要以多种木质素降解酶协同作用完成的。因此,下一步还需构建表达木质素过氧化物酶、锰过氧化物酶、多功能过氧化物酶的工程菌,并进行工程菌优化组合研究;还可以通过优化酶基因关键调控元件(提高启动子强度、优化信号肽),改造和优化工程菌代谢通路等研究来进一步提高酶蛋白产量。
