HPLC法测定注射用盐酸头孢替安的有关物质
2022-03-24闫灵锐费小琴周艳婷
王 庆,闫灵锐,费小琴,周艳婷
重庆圣华曦药业股份有限公司,重庆 401336
盐酸头孢替安是诺华制药和日本武田共同研制开发的第二代头孢类广谱抗生素,对革兰氏阳性菌和阴性菌感染均有效。临床上不仅用于成人多种感染的治疗[1-4],还用于儿童肺炎[5-6]、肠炎[7]、扁桃体炎[8-9]、猩红热的治疗[10]及围术期感染的预防[11]。以无水碳酸钠为辅料的注射用盐酸头孢替安原研品,最早于1980年10月在瑞士获批,并在1981年2月,以商品名Halospor在日本上市。2002年4月,诺华制药将销售权转移给富山化学。2019年4月,富山化学开始自行生产和销售。原研品主要上市国为日本、荷兰、澳大利亚,目前国内尚未进口。
注射用盐酸头孢替安收载于《美国药典》43版(USP43)[12]和《日本药典》17版(JP17)[13],但均未收载有关物质的检查项。头孢类产品的有关物质是关系到产品是否安全有效、不良反应多寡,甚至患者生命安全的重要质量控制指标。国内对有关物质方法学研究的报道较少。周冉等[14]采用与USP43中含量测定方法相同的等度洗脱高效液相色谱法(high performance liquid chromatography,HPLC)进行检测,该方法主峰出峰时间早,能分离的杂质有限;王珵等[15]在USP43已有等度洗脱的基础上,优化为梯度冲洗,但是未采用对照品对分离度进行验证,专属性中未对主峰的纯度进行检测;田冶等[16]进行了注射用盐酸头孢替安中杂质的质谱定性研究,同时提供了定性的液相条件,但在供试品高温降解的条件下,降解杂质和主峰的分离度较低。
本研究根据文献中报道的注射用盐酸头孢替安杂质[17-21],结合原料药的合成工艺[22-23],确定注射用头孢替安的主要杂质为杂质A~I,结构式见图1;并建立了完全分离杂质、可稳定重复的有关物质的检测方法,为控制盐酸头孢替安的质量提供依据。
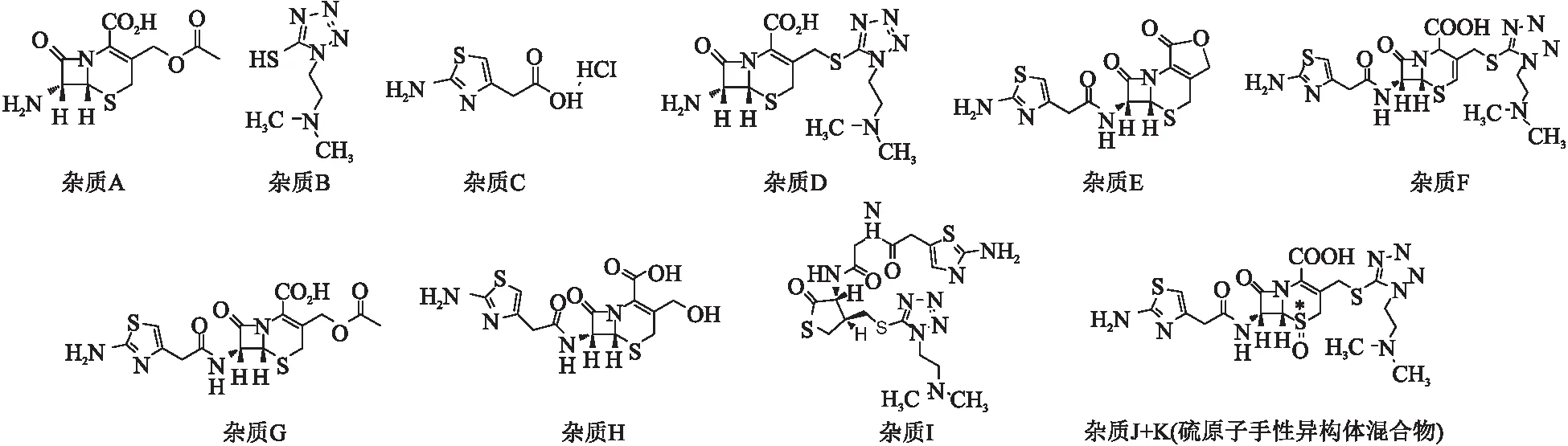
图1 杂质A~I的化学结构
1 仪器与试药
1.1 仪器
LC-2030C液相色谱仪(带二级管阵列检测器,日本岛津科技有限公司);PHS-3W pH计(上海班特仪器制造有限公司);XS105DU电子天平[梅特勒-托利多国际贸易(上海)有限公司];LB-Solution色谱工作站(日本岛津科技有限公司);Eco-S15实验室纯水系统(上海和泰仪器有限公司)。
1.2 试药
盐酸头孢替安对照品(批号130565-201703,中国食品药品检定研究院);盐酸头孢替安对照品(批号P42-181207G-R-1,重庆圣华曦药业股份有限公司);注射用盐酸头孢替安[规格1.0 g,批号190501(验证用),批号05200101Y、05200102Y、05200103Y(检验用),辅料无水碳酸钠,重庆圣华曦药业股份有限公司];杂质A(批号M347-181001A-R,重庆圣华曦药业股份有限公司);杂质B(批号M410-181001A-R,重庆圣华曦药业股份有限公司);杂质C(批号180301,重庆常捷医药有限公司);杂质D(批号P42I1-190301G-R,重庆圣华曦药业股份有限公司);杂质E(批号P42I2-180301G-R-1,重庆圣华曦药业股份有限公司);杂质F(批号TBTAF2018080101,湖南青纯科技有限公司);杂质G(批号P42I3-190701-R,重庆圣华曦药业股份有限公司);杂质H(批号20180329,深圳卓越生物科技有限公司);杂质I(批号20190322,深圳卓越生物科技有限公司);杂质J、K混合物(批号P42I4-190701-R,重庆圣华曦药业股份有限公司);乙腈(批号T7QA1H,霍尼韦尔中国有限公司);磷酸二氢钾(批号190717,重庆博艺化学试剂有限公司);磷酸氢二钠(批号2019020101,成都市科隆化学品有限公司)。
2 方法与结果
2.1 色谱条件
色谱柱:NanoChrom ChromCore AQ C18column(250 mm×4.6 mm,5 μm);检测波长:254 nm;柱温:40 ℃;流速:1.2 mL·min-1;进样量:20 μL。流动相A:0.05 mol·L-1磷酸二氢钾缓冲液(用0.05 mol·L-1磷酸氢二钠溶液调节pH至4.9);流动相B:0.05 mol·L-1磷酸二氢钾缓冲液(用0.05 mol·L-1磷酸氢二钠溶液调节pH至4.9)-乙腈(80∶20)。梯度程序:按照表1进行洗脱。
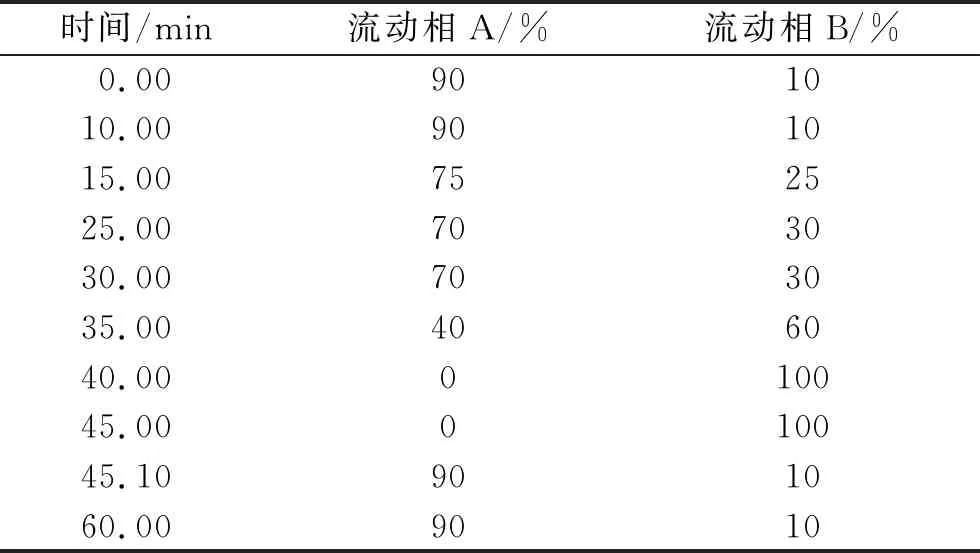
表1 梯度洗脱程序
2.2 溶液配制
空白:稀释剂。杂质A、E储备液:分别取杂质A、E对照品适量,加2 mol·L-1盐酸溶液约1 mL溶解,加稀释剂稀释成每1 mL中含杂质A、杂质E 1 mg的溶液。杂质B、C、D储备液:分别取杂质B、C、D对照品适量,加稀释剂稀释成每1 mL中含杂质B、C、D 1 mg的溶液。系统适用性溶液:取盐酸头孢替安对照品适量,加稀释剂稀释成每1 mL中含头孢替安各1 mg的混合溶液。量取系统适用性溶液1 mL与杂质A~E储备液各1 mL,加稀释剂稀释成每1 mL中含杂质A~E、头孢替安约10 μg的混合溶液。供试液:精密称定本品适量,加稀释剂溶解,制成每1 mL中约含头孢替安1 mg的溶液(临用前新制)。对照液:精密量取供试品溶液适量,用稀释剂稀释成每1 mL中约含头孢替安10 μg的溶液。
精密量取空白、供试液、对照液各20 μL,注入液相色谱仪,记录色谱图。
2.3 系统适用性要求
精密量取系统适用性溶液20 μL,注入液相色谱仪,记录色谱图,见图2。杂质B、C、D、A、E和头孢替安依次出峰,各已知杂质之间、各已知杂质与头孢替安峰之间的分离度均不得小于1.5。
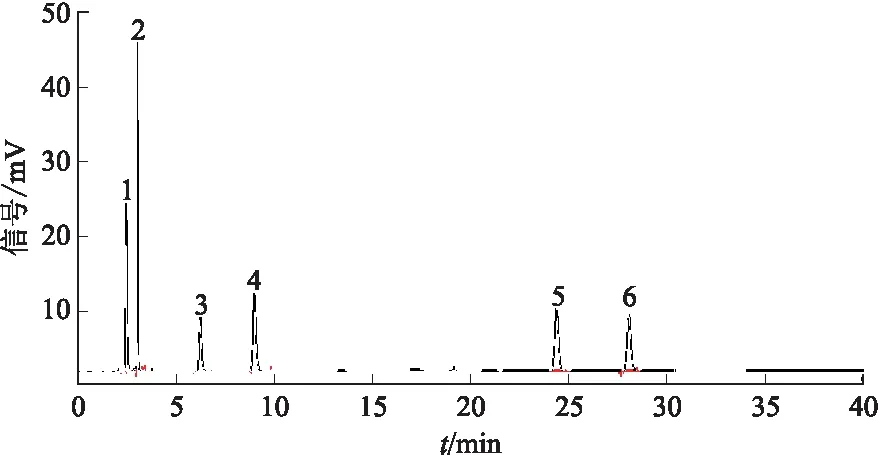
注:1.杂质B;2.杂质C;3.杂质D;4.杂质A;5.杂质E;6.头孢替安。
2.4 方法学验证
2.4.1专属性实验 杂质A~K储备液:分别取杂质A~K对照品适量,溶解后,加稀释剂稀释成每1 mL中含杂质A~K 1 mg的溶液。混合溶液:取盐酸头孢替安对照品适量,加杂质储备液,用稀释剂稀释制成每1 mL含各杂质10 μg、头孢替安1 mg的溶液。空白辅料溶液:取处方量无水碳酸钠,同2.2项下供试品溶液配制方法制备,即得。降解溶液:取供试品适量,分别进行光照、酸、碱、氧化和高温降解,酸、碱降解中和,按2.2项下方法配制供试品溶液。含量测定溶液:分别精密量取上述各种降解有关物质溶液稀释5倍,进行含量测定。取上述溶液进样,记录色谱图。结果见表2。
结果表明,辅料不干扰有关物质的检测。降解条件下,主峰与前、后杂质可以有效分离,且峰纯度符合要求,物料平衡在《中华人民共和国药典》2015年版规定的范围内。强制降解结果还表明,本品易在强酸、强碱、高温、氧化条件下发生降解;酸、碱降解的主要产物为杂质B、杂质E和杂质H;氧化降解的主要产物为杂质J与杂质K;高温降解的主要产物为杂质B、杂质E和杂质F。结果见图3。
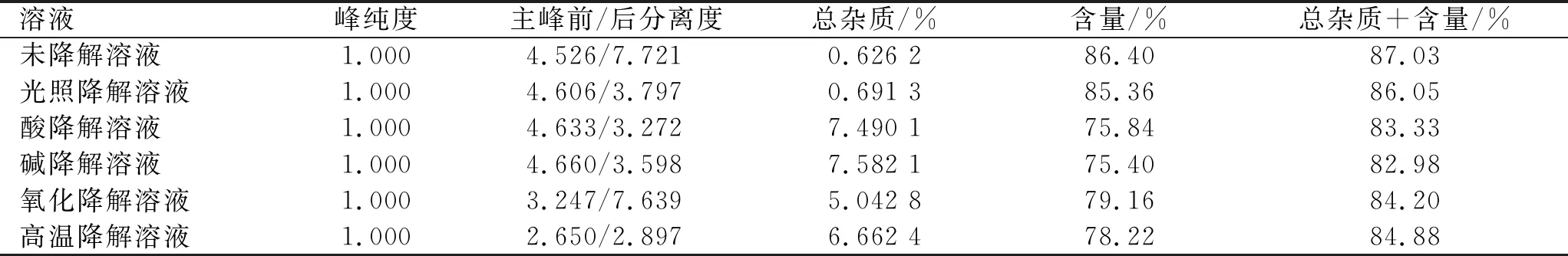
表2 专属性实验结果
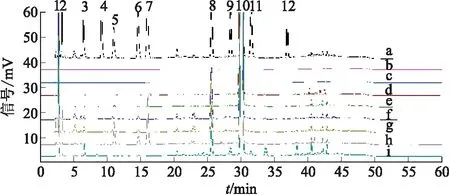
注:a.杂质混合溶液;b.空白溶液;c.空白辅料溶液;d.未降解溶液;e.光照降解溶液;f.酸降解溶液;g.碱降解溶液;h.氧化降解溶液;i.高温降解溶液;1.杂质B;2.杂质C;3.杂质D;4.杂质A;5.杂质J;6.杂质H;7.杂质K;8.杂质E;9.杂质F;10.头孢替安;11.杂质I;12.杂质G。
2.4.2检测限与定量限实验 储备液:分别取杂质A~K、盐酸头孢替安对照品适量,加稀释剂制成每1 mL含杂质0.2 mg的溶液;取储备液适量,逐步稀释制备检测限(3倍噪音质量浓度)和定量限(10倍噪音质量浓度)的实验溶液,实验结果见表3。各杂质和头孢替安的检测限质量浓度低于限度质量浓度10%,定量限不超过限度质量浓度的30%,符合检测要求。
2.4.3线性关系 精密称取杂质A~E及盐酸头孢替安对照品适量,稀释后配制成限度分别为10%、20%、50%、80%、100%、120%、160%的混合溶液。取线性溶液各20 μL分别注入液相色谱仪,记录色谱图,并以峰面积为x轴、质量浓度为y轴,进行线性回归,结果见表4。结果显示,各成分在10%~160%的范围内,将峰面积与质量浓度进行线性回归计算,相关系数均大于0.999;响应因子的相对标准偏差均小于10%,表明各组分的线性关系良好。
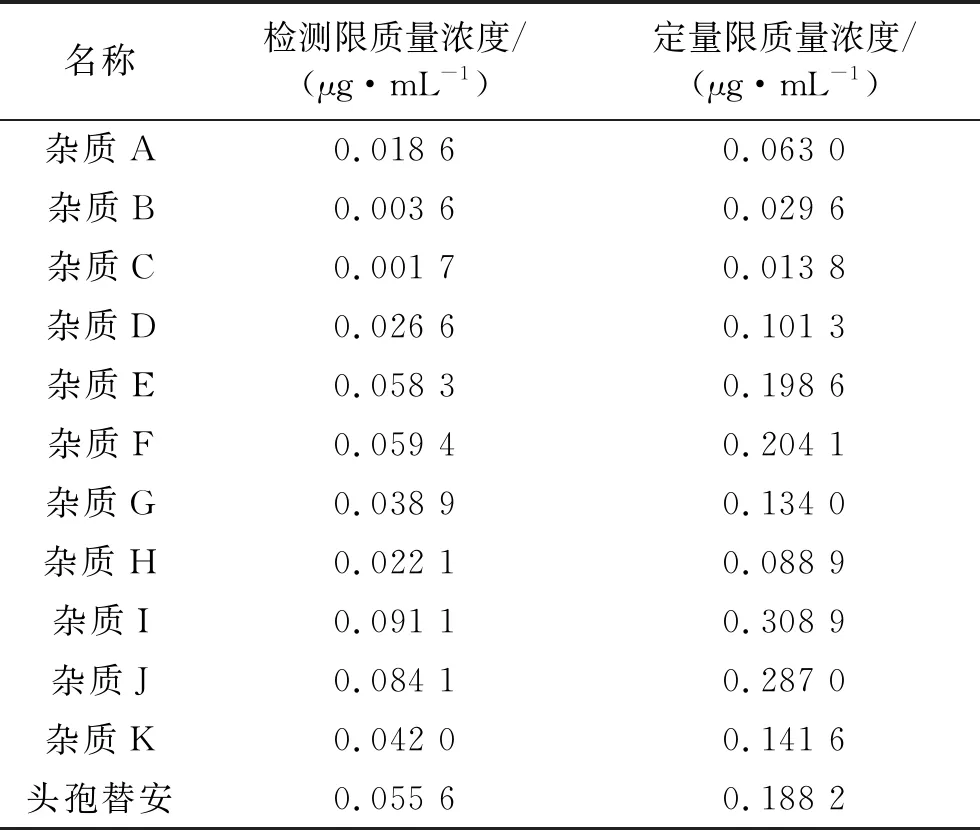
表3 检测限与定量限测定结果
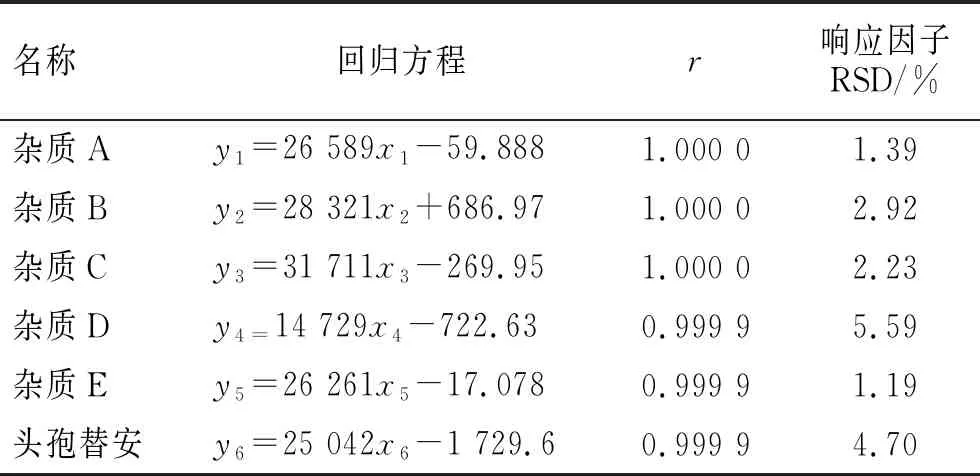
表4 线性关系实验结果
2.4.4精密度实验 杂质储备液:分别取杂质A~E适量,精密称定,加稀释剂配制成每1 mL含杂质A~C、杂质E 0.2 mg、杂质D 0.3 mg的溶液。本底供试液:取盐酸头孢替安对照品适量(相当于头孢替安约50 mg)和无水碳酸钠12.1 mg,精密称定,置于50 mL量瓶中,用稀释剂溶解并稀释至刻度,摇匀。准确度50%供试液:取盐酸头孢替安对照品适量(相当于头孢替安约50 mg)、无水碳酸钠12.1 mg,精密称定,各3份,分别置于3个50 mL量瓶中,精密加入杂质储备液A~E各0.25 mL,杂质储备液B 1.25 mL,加稀释剂溶解并稀释至刻度,摇匀。准确度100%供试液:精密加入杂质储备液A~E各0.5 mL,其他与50%供试液的制备方法相同。准确度150%供试液:精密加入杂质储备液A~E各0.75 mL,其他与50%供试液的制备方法相同。
回收率测定结果见表5。由表5可见,各质量浓度下杂质A、B、C、D、E的平均回收率分别为98.91%、99.38%、96.36%、97.80%、98.31%,均在90%~108%范围内,且回收率的RSD值分别为3.07%、0.84%、1.70%、2.71%、4.18%,均小于10%,即杂质A、B、C、D、E的准确度符合要求。
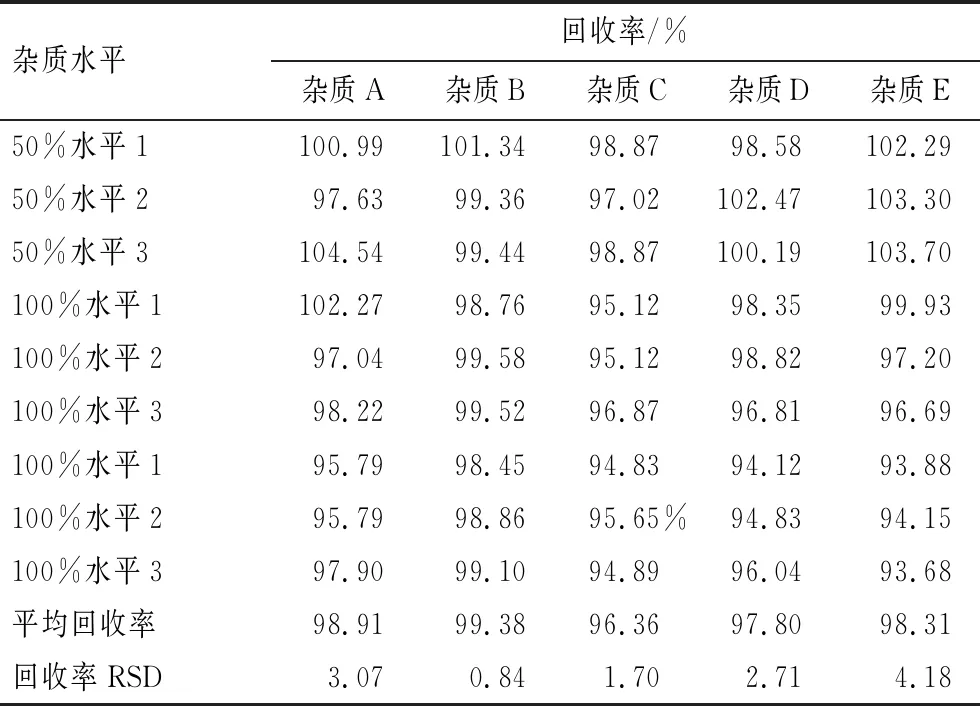
表5 回收率实验结果
2.4.5重复性实验 杂质储备液:按照2.4.4项下杂质储备液的配制方法制备。取注射用盐酸头孢替安,按照2.4.4项下准确度100%供试液的配制方法制备供试品溶液6份,分别进样分析,计算已知杂质RSD值及总杂质RSD值。测得数据均符合要求,RSD值(n=6)均小于10%,说明该方法的重复性较好。
2.4.6中间精密度 按照2.4.3项下准确度100%供试液的配制方法制备供试品溶液6份,不同时间、不同仪器、不同人员进行实验,测得数据均符合要求,RSD值(n=6、n=12)均小于10%,结果见表6,说明该方法的中间精密度较好。
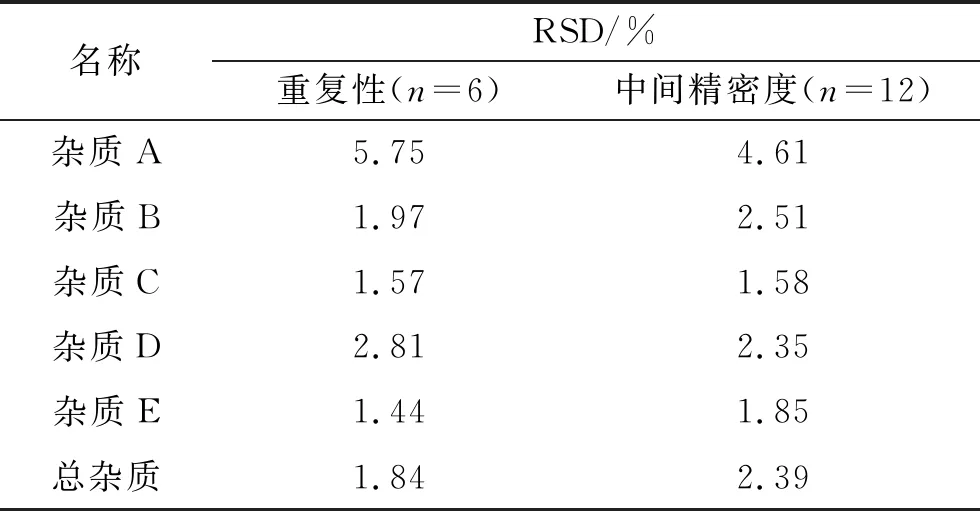
表6 重复性与中间精密度实验结果
2.4.7溶液稳定性考察 取同一份供试品溶液适量,置于2个进样小瓶,分别于室温和4 ℃放置,于0、1、2、4、6、8 h进样检测,结果显示室温溶液在2 h时总杂质相比0 h大1.1%;4 ℃放置,8 h时总杂质增加0.17%,2 h总杂质增加0.077%,表明该溶液在室温下不稳定,在4 ℃条件下放置2 h内基本稳定。
2.4.8耐用性考察 考察不同条件(pH±0.1、波长±2 nm、柱温±2 ℃、磷酸盐浓度±0.005 mol·L-1、流速±5%、不同批次色谱柱)下,系统适用性及样品检测结果。结果表明,在上述条件下系统分离度合格,测得本品杂质A~E的含量及总杂质与拟用条件的绝对差值均在±0.1%范围内,表明该方法的耐用性良好。
2.4.9样品测定 检测注射用盐酸头孢替安3批,批号分别为05200101Y、05200102Y、05200103Y(1.0 g),结果见表7。
3 讨论
3.1 检测波长的确定
本品USP43和JP17均未收载有关物质,2个标准含量项目的检测波长均为254 nm。通过检测各有关物质的紫外吸收波长,多数有关物质峰与主峰在254 nm处有明显且相似的吸收,故选择254 nm作为检测波长。
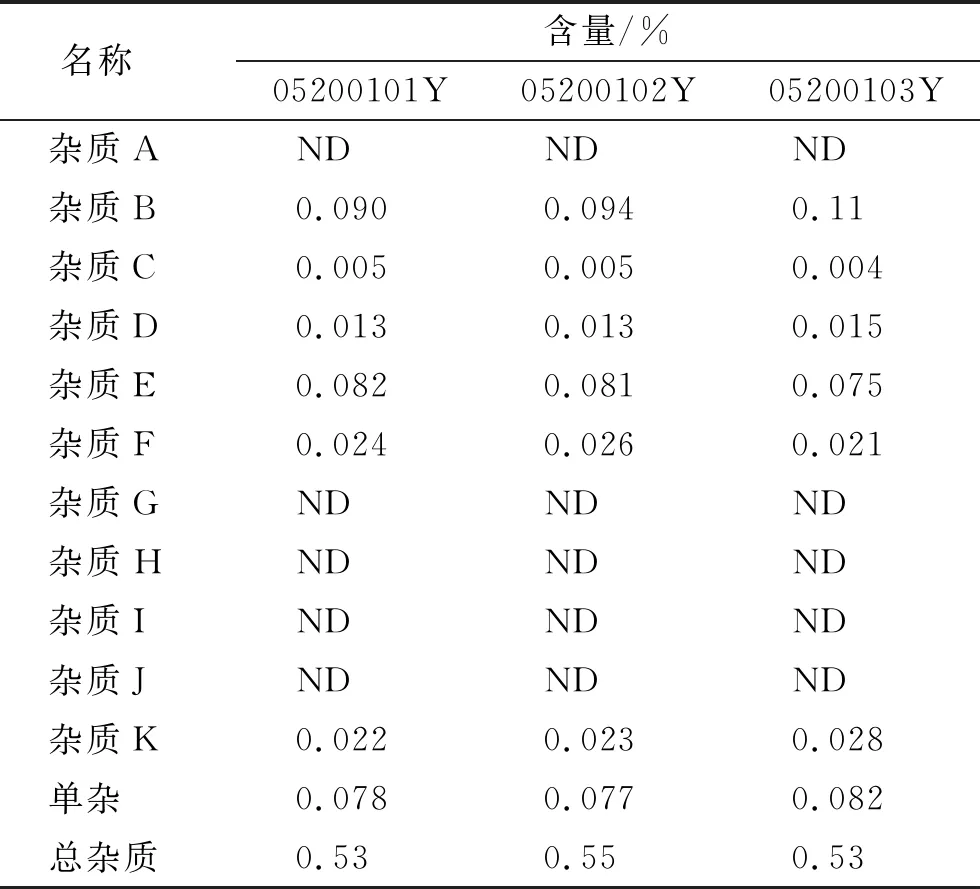
表7 样品测定结果
3.2 pH的选择
考察了不同pH(4.2、4.9、5.6)流动相的分离效果,各杂质与头孢替安的出峰时间均随pH升高而延后;pH升高,杂质K与杂质H、杂质E与杂质F分离度上升,但杂质H与杂质J、杂质G与杂质I分离度会下降,根据主成分与杂质混合溶液色谱图,在pH4.9时分离度整体较好,在pH5.6时出峰时间太晚,因此选择流动相pH为4.9。
3.3 柱温的选择
考察了不同柱温(30、35、40 ℃)流动相的分离效果,升高柱温,有利于杂质H与杂质K、杂质I与头孢替安的分离。柱温在40 ℃时,各杂质的分离度良好,因此最终确定柱温为40 ℃。
4 结语
杂质A、C、D为工艺杂质在产品中残留的可能性大,杂质B、E为主要降解杂质,因此,本研究将杂质A、B、C、D、E作为主要已知杂质,采用以系统溶液进行定位,加校正因子的自身对照法进行控制;对于杂质F~K,采用不加校正因子的自身对照法进行控制。经过对波长、pH、柱温、梯度程序等条件的优化,本研究建立了一种专属性、准确性、耐用性均较好的方法。方法学验证结果表明,该方法适用于注射用盐酸头孢替安杂质含量的检测与控制。
