大豆长片段插入/缺失标记的开发与应用
2022-03-22史晓蕾刘志芳孟庆民杨春燕王冬梅张孟臣
李 曼,史晓蕾,邸 锐,刘志芳,孟庆民,付 才,杨春燕,王冬梅,张孟臣,张 洁,闫 龙
(1.华北作物改良与调控国家重点实验室,河北省植物生理与分子病理学重点实验室,河北农业大学 生命科学学院,河北 保定 071001;2.河北省农林科学院 粮油作物研究所,国家大豆改良中心石家庄分中心,农业农村部黄淮海大豆生物学与遗传育种重点实验室,河北省作物遗传育种实验室,河北 石家庄 050035;3.河北省种子总站,河北 石家庄 050031;4.河北省农林科学院 遗传生理研究所,河北 石家庄 050051)
大豆(Glycinemax(L.)Merrill)为人类提供了1/3的植物蛋白和食用油,在农产品贸易中地位重要。近年来,我国大豆种业市场日趋活跃,大豆种子企业初具规模,育成品种(如黑河43、齐黄34、中豆63等)也已经实现品种权高价转让。但种子纯度低、套名装袋等不规范行为在市场上屡有发生。大豆种质资源保存过程中,同名异种、同种异名等现象也时有发生[1]。
我国各级区域试验要求利用分子标记鉴定种子纯度、并建立品种指纹图谱[2-3]。简单重复序列(Simple Sequencing Repeat,SSR)标记[4]、单核苷酸多态性(Single Nucleotide Polymorphic,SNP)标记[5]和插入/缺失片段长度多态性(Insertion/Deletion,InDel)标记[6-8]等,可用于大豆品种纯度检测和指纹图谱构建[9]。SSR标记是目前各级区试品种纯度检测中普遍使用的分子标记,但利用SSR标记进行基因型分析时,多数位点产生的等位变异数目多,导致不同批次试验结果间可比性差。插入/缺失标记是由于在不同个体间,等位基因位点的DNA序列发生核苷酸片段的插入/缺失产生的多态性变异[10-11],具有分布广、全基因组密度高、等位变异数量少等优点。当某个标记的插入/缺失片段长度达到一定阈值后,其等位变异差异即可以通过PCR反应和琼脂糖凝胶电泳分辨,从而开发成稳定可靠、操作简便的分子标记。
为鉴定黄淮海地区各育种单位育成品系的农艺性状、增产潜力以及适应区域[12],自2010年以来,黄淮海大豆育种协作网对有关育种单位选育出的新品系在西北春大豆区、黄淮夏大豆区覆盖的10个省市开展了联合多点鉴定,共有1 380份品种(含对照)参加联合试验。多点鉴定试验报告已经成为推荐参加黄淮海地区各省级区试和国家区试主要的试验依据。
本研究旨在开发在基因组中均匀分布、扩增效果好、插入/缺失片段大、可用于大豆品种指纹图谱构建、操作简便的插入/缺失标记,并利用所开发的标记,构建2018年黄淮海联合鉴定的96份大豆参试材料的指纹图谱,进行遗传多样性分析,为大豆种质资源评价利用和品种纯度鉴定等提供参考。
1 材料和方法
1.1 试验材料
1.1.1 用于开发InDel标记的试验材料 用于开发InDel标记的材料包括:一年生野生大豆ZYD02738,美国引进品种Hobbit,我国东北地区育成品种绥农14、吉育101,黄淮海地区育成品种(系)冀豆17、冀豆12、冀HJ117、中黄13、中黄42、齐黄34、郑196、徐豆16。
1.1.2 用于遗传多样性分析的鉴评材料 从2018年大豆黄淮海多点联合鉴定试验材料中,按照选育单位来源,选取96份材料,构建指纹图谱并进行遗传多样性分析。
1.2 试验方法
1.2.1 基因组DNA提取 按照康为世纪Nuclear Plant Genomic DNA Kit 新型植物基因组DNA提取试剂盒提取参试大豆材料的DNA。收集提取的DNA溶液后,用Nanodrop分光光度计检测样品DNA浓度和质量(OD260/OD280≈1.8),将检测合格的DNA样品浓度调至20 ng/μL,-20 ℃保存备用。
1.2.2 大豆基因组重测序 提取植物材料DNA利用超声波将质检合格的植物材料DNA片段化,然后将片段化的DNA进行片段纯化、末端修复、3′端加A、链接测序接头,用琼脂糖凝胶电泳选择片段长度大小,PCR扩增建立测序文库,用Illumina HiSeqTM2500对质检合格的文库进行10倍深度重测序。对测序得到的原始reads进行质量评估,过滤得到Clean reads,用于后续的基因组序列拼装[13-14]。重测序由北京诺禾致源科技有限公司完成。
1.2.3 InDel位点挑选与引物设计 将12份材料重测序结果与参考基因组序列(Glyma.Wm82.a2v1)进行对比,使用GATK软件[15],挖掘大豆基因组中的InDel标记。在此基础上,挑选长片段插入/缺失位点设计引物,选择标准为:在大豆每条染色体选择均匀分布的位点;插入/缺失片段长度≥20 bp;每个位点有且只有2个等位变异;多态性信息含量大于0.25。利用BioXM 2.6[16]设计引物,由金唯智公司合成。
1.2.4 PCR反应体系与引物筛选 PCR扩增采用康为世纪公司的2×EsTaqMasterMix(Dye),20 μL PCR反应体系为:DNA(20 ng/μL)1 μL,2×EsTaqMasterMix 10 μL,上游、下游引物(10 μmol/L)各1 μL,ddH2O 7 μL,将各组分充分混匀后,置于PCR仪中,反应程序为:94 ℃预变性3 min;94 ℃变性15 s,55 ℃退火15 s,72 ℃延伸 30 s,34个循环;72 ℃终延伸10 min。取5 μL PCR产物在1.0%琼脂糖凝胶进行电泳检测。120 V电泳35 min,凝胶成像仪下观察。能够扩增出条带稳定、多态性高、且可用肉眼观察插入/缺失类型条带的引物可作为分子标记加以应用。
1.3 数据分析
1.3.1 InDel位点检测注释与分析 使用SnpEff软件[17]对InDel进行注释,统计分析基因间隔区、外显子、内含子等区域内分布的InDel标记的数量。
1.3.2 遗传多样性指数计算 利用Popgene 32[18]软件对引物在供试材料的扩增结果进行分析,计算每对引物的遗传多样性指数。
期望杂合度(Expected heterozygous number)[19]:即基因多样性He=1-∑Pi2,其中,Pi为第i个等位基因的频率,计算每个位点的期望杂合度,取平均值代表该群体的期望杂合度;
Nei指数(Nei diversity index)[19]:Nei=nHe/(n-1),其中,n为样品数,He为期望杂合度;
香农指数(Shnnon wiener index)[19]:SHI=-∑(Pi×lnPi),其中Pi为第i个等位基因的频率,分别计算获得各位点的SHI,取平均值代表该群体的SHI;
多态性信息含量(Polymorphysm information content,PIC)[19]:
其中Pi和Pj分别为第i个和第j个等位基因频率,n为等位基因数。
1.3.3 Structure群体遗传结构分析 利用基于贝叶斯法的Structure 2.3.4软件[20]对96份材料进行遗传结构分析。步骤如下:①利用Excel整理电泳结果,变异类型为插入的位点记为1,变异类型为缺失的位点记为0,未检测到变异类型的位点记为9,将Excel转换为Structure支持的格式。②确定最佳群体组群K值。软件K值范围设置在2~7,重复次数设置为2,每个参数运行10次,每次运行的Burnin time和重复次数都设置为80 000。依据后验概率值LnP(D)确定分组K值,当LnP(D)值最小时的K值即确定为最佳K值。
1.3.4 指纹图谱构建 根据每个InDel标记对试验材料检测出等位变异的类型及有无构建指纹图谱,变异类型为插入的位点记为1,变异类型为缺失的位点记为0,未检测到变异类型的位点记为9,品种(系)纯度计算公式:P=(S1-S2)/S1×100%,其中,S1为检测InDel位点数,S2为杂合位点数[2,21]。
2 结果与分析
2.1 插入/缺失片段分布特征
参试材料全基因组重测序结果显示,正确识别率大于Q20(99.9%)的碱基占比平均值为97.50%,正确识别率大于Q30(99.9%)的碱基占比平均值为93.48%,基因组GC含量平均值为36.23%,样品平均重测序覆盖率为98.70%,平均测序深度18.73X。
检测到SNP、InDel共2种主要变异类型。其中,检测到插入/缺失片段长度大于20 bp(包括20 bp)的InDel标记的数量为66 561,平均每条染色体的InDel标记的数量为3 262,其中第18号染色体分布的InDel标记最多,数量为4 521,第12号染色体分布的InDel最少,数量为2 019。各品种InDell数量不同(图1),野生大豆基因组内分布的InDel多,大豆品种Hobbit染色体内分布的InDel较少。

A—L.冀豆17、冀豆12、绥农14、中黄13、冀HJ117、Hobbit、吉育101、齐黄34、徐豆16、ZYD2738、郑196、中黄42。A—L.Jidou 17,Jidou 12,Suinong 14,Zhonghuang 13,JiHJ117,Hobbit,Jiyu 101,Qihuang 34,Xudou 16,ZYD2738,Zheng 196,Zhonghuang 42.
如表1所示,检测到的66 561个InDel标记主要位于基因间隔区、内含子区、外显子区、5′-UTR、3′-UTR、基因上游序列和下游序列。位于基因上游序列和下游序列的InDel数量分别为17 190个(25.83%)和13 602个(20.44%),位于基因间隔区的InDel数量为22 893个(34.39%),位于外显子的InDel数量为125个(0.19%),位于内含子的InDel数量为8 222个(12.35%),位于3′-UTR和5′-UTR的InDel数量分别为1 004(1.51%),617个(0.93%)。

表1 插入/缺失在基因组的分布Tab.1 Distribution of InDel in the genome
检测到的66 561个InDel标记中,可鉴定长度的InDel有65 229个。片段长度大小介于20~40 bp的InDel数量为42 453个,介于41~60 bp的InDel数量为13 044个,介于61~80 bp的InDel数量为5 034个,介于81~100 bp的InDel数量为2 285个,大于100 bp的InDel数量为2 413个(图2)。
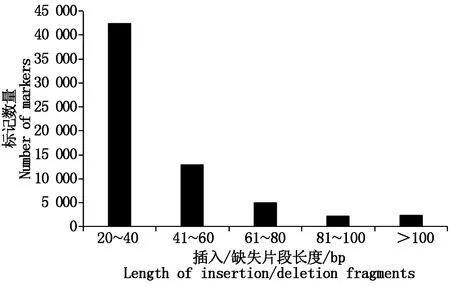
图2 插入/缺失片段的长度分布特征Fig.2 Length distribution characteristics ofinsertion/deletion fragments
分析66 561个InDel标记在12份重测序材料中的PIC指数、Nei多样性指数、香农指数可得,PIC指数为0.067~0.375,Nei多样性指数标记数量为0.710~0.667,香农指数在0.154~0.693(图3)。其中,PIC指数集中分布在0.100~0.200,0.300~0.400,所占比例分别为35.58%,33.01%;Nei多样性指数集中分布在0.100~0.200,所占比例为33.56%;香农指数集中分布在0.200~0.300,0.600~0.700,所占比例分别为29.30%,24.09%。
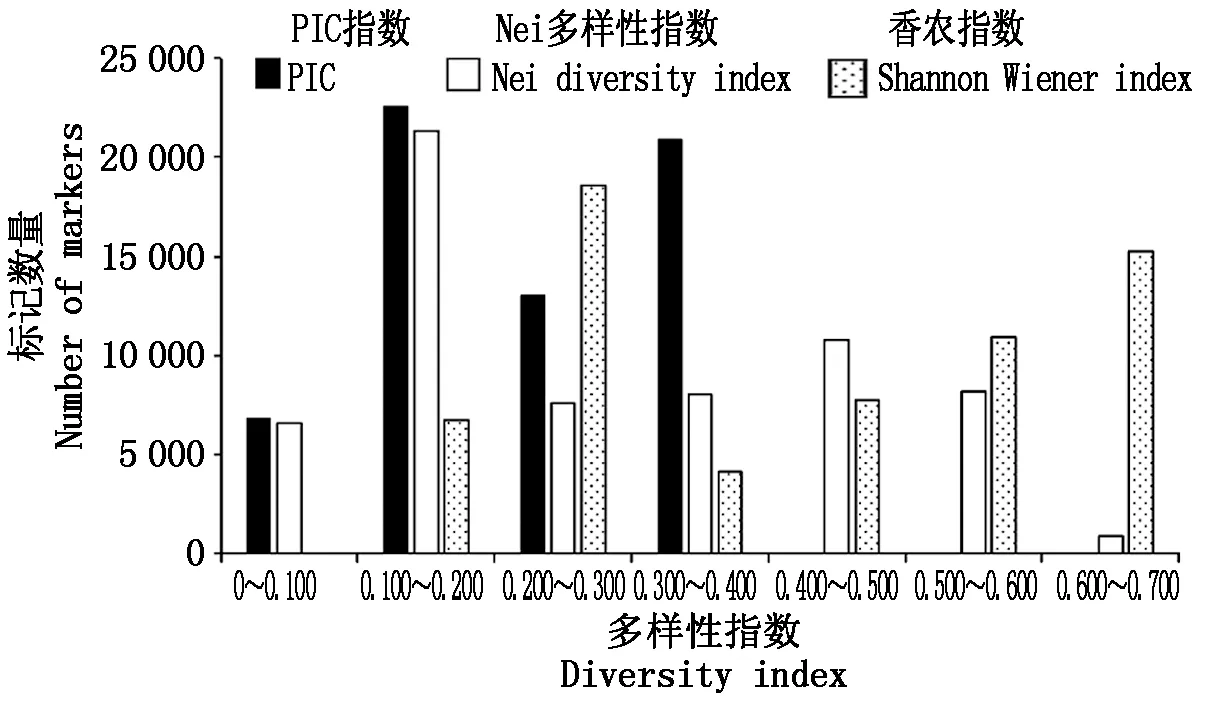
图3 InDel标记的PIC指数、Nei多样性指数、香农指数分布特征Fig.3 PIC,Nei diversity index,Shannon Wiener index′s distribution characteristics of InDel marker
2.2 长片段InDel标记开发
共设计160对InDel标记引物,在55 ℃的退火温度下,109对引物扩增效果稳定、条带清晰。其中,32对引物的扩增条带经1%琼脂糖凝胶电泳后,肉眼可分辨片段插入/缺失类型(表2、图4)。
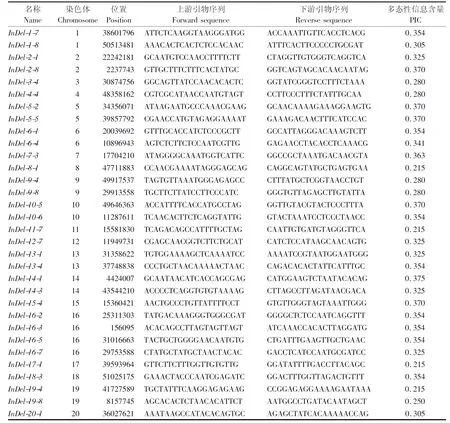
表2 32对插入/缺失标记引物信息Tab.2 Primer information of 32 insertion/deletion markers
2.3 InDel标记应用
利用 Popgene 32 软件对32对引物在96份大豆材料中的扩增结果进行分析,每对引物的Nei指数分布在0.148 6~0.499 9,均值为0.393 5;香农指数分布在0.280 7~0.693 1,均值为0.577 9;PIC指数分布在0.152 8~0.595 3,均值为0.396 3,表明InDel标记在96份大豆材料中能较好地反映出其丰富的遗传多样性信息。
用InDel标记对参试材料构建指纹图谱时,将检测出变异类型为插入的位点记为1,变异类型为缺失的记为0,未检测到变异类型的位点记为9(表3)。结果发现,用32对引物可将96份材料区分开,参试的大豆材料纯度为96.84%,每份材料有唯一的指纹代码,且没有同物异名现象发生,因此,证明基于InDel标记构建的遗传图谱可以用于品种的鉴定。

表3 96份黄淮海多点鉴定试验材料指纹图谱Tab.3 Fingerprint of 96 Huanghuaihai test materials
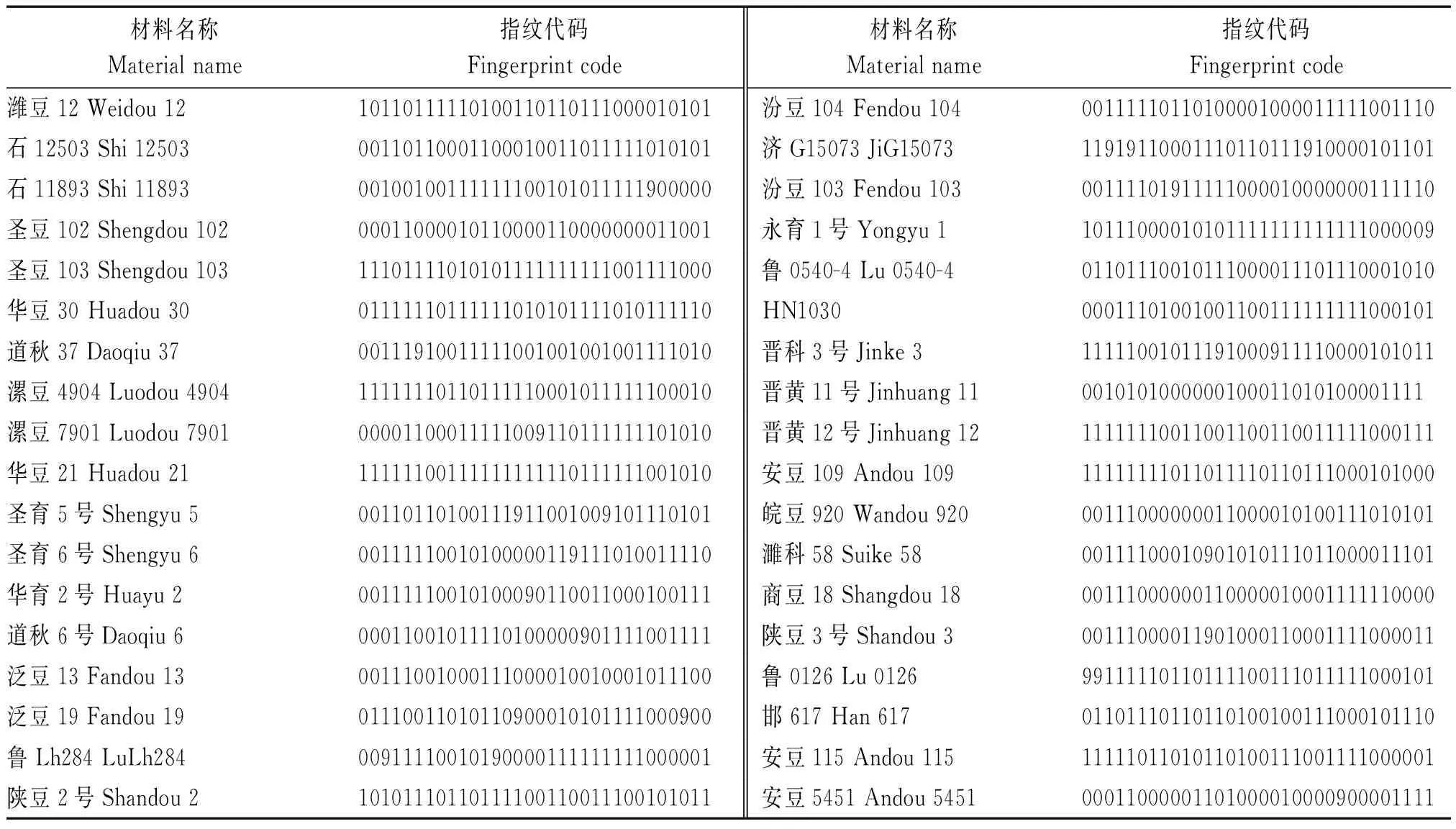
表3(续)
群体遗传结构分析显示,当K=5时,ΔK达到最大值。当K=5时拟合结果中的bar plot绘制大豆各品种所占比例。从图5可以看出,Structure软件将96份大豆材料分成了五大类群,分别用5种颜色表示,柱状图的高度描述的是不同品种被分到五类群所占比例。

图5 96份大豆种质的遗传结构Fig.5 The genetic structure of 96 soybean germplasms
3 结论与讨论
本研究中,InDel标记在基因组中的分布,呈现出两端分布密度高,靠近着丝粒区域分布密度低的趋势。并且,基因内无转录信息区域分布的InDel标记最多(34.39%),基因上、下游序列分布的InDel次之(分别为25.83%,20.44%),基因片段内分布的InDel最少(0.19%)。究其原因,如果InDel分布在基因CDS区内,核苷酸片段的插入/缺失可能会导致基因功能的丧失,进而影响生物体正常生命活动,导致该InDel标记不能在物种内稳定存在;反之,如果InDel发生在无转录信息的区域,则该InDel标记在物种中保留下来的机会将大大提高。
InDel标记具有特异性高、稳定性好、多态性高、经济实用等特点,因此,InDel标记对种质资源的遗传多样性及种质资源鉴定具有广阔的研究前景[22],在大豆[23]、水稻[24]、玉米[25]等作物中应用较多。本研究所开发的长片段插入/缺失标记具有2个突出特点:一是在种质资源里,每个位点有且仅有2个等位变异,因此,检测结果易于读取,不同批次试验结果也易于比较。而目前国家区域试验品种纯度分析采用30对SSR标记进行检测[2],大豆中平均每个SSR位点等位变异在6个左右,增加了数据读取的难度。二是插入/缺失片段长(大于40 bp),可通过琼脂糖凝胶电泳检测。前人[26]开发的插入/缺失标记,没有专门关注片段长度问题,大多数是介于1~5 bp,需要通过聚丙烯酰胺凝胶电泳检测,操作繁琐、不便于检测。本研究开发的标记,可用琼脂糖凝胶电泳对标记的有效性进行检测,检测手段方便快捷,结果便于观察分析,可靠性高。利用本研究开发的InDel标记,构建了2018年黄淮海大豆多点鉴定参试品系DNA指纹图谱,参试的大豆材料纯度为96.84%,没有同物异名现象发生,进一步证实了所开发标记的可靠性和简便性。
本研究以全基因组重测序技术为基础,对12份大豆种质资源进行重测序,建立了数据丰富、准确性高的测序文库。比对大豆参考基因组序列,设计开发基于电泳检测技术可清晰分辨的长片段插入/缺失InDel标记,可以对大豆种质进行遗传多样性分析,具有良好的应用前景。
