脂蛋白肾病1例并文献复习
2022-03-21王雨晨严跃红李翠翠郎士娟
王雨晨,严跃红*,李翠翠,郎士娟
(1.广州医科大学附属第五医院肾内科,广东广州 510700;2.广州医科大学附属第五医院康复治疗科,广东广州510700)
脂蛋白肾病(lipoprotein glomerulopathy,LPG)是一种遗传性肾脏疾病,其发病率较低,有研究者根据现有数据库信息,估计LPG在中国的发病率约为1.43/10万[1]。其发病机制尚未明确,目前主要认为由血浆载脂蛋白E(apolipoprotein E,ApoE)[2-3]的变异导致脂代谢异常引起。LPG 缺乏特征性临床表现,临床上主要以肾病综合征为主要特征,常伴有高脂血症,但未有合并其它脏器脂质代谢异常的损伤表现的病例报道。其诊断主要依赖于组织病理学,具体表现为肾小球毛细血管管腔扩张并在其内存在大量嗜酸性脂蛋白血栓,油红O染色呈阳性[2]。LPG目前尚无有效的根治方法,临床上主要以降脂、护肾、减少蛋白尿等对症治疗为主。由于缺乏针对性的治疗,LPG患者预后往往差强人意。大约50%的LPG患者在发病后20余年内进展为终末期肾病[4]。在接受肾移植的患者中,随访5个月至2年复发[5]。在中国,成人脂蛋白肾小球疾病病例很少有报道,在此,我们报道一例33岁男性,无已知肾脏疾病家族史,因“排泡沫尿5月余”为主诉入院。并对既往国内外脂蛋白肾小球肾病相关文献进行复习。
1 病例资料
患者黄某,男,33岁,因“排泡沫尿5月余”为主诉于2022年2月9日入院。患者于5个月前无明显诱因出现尿中泡沫,无肉眼血尿,无尿量减少,完善相关检查:20221-11-24 尿蛋白 4+,尿红细胞 4+,肌酐3.00 umol/L,尿酸 461 umol/L,2021-11-24 肾脏彩超提示:双肾实质回声稍增强,予以护肾、降尿蛋白、降尿酸等对症处理,期间多次复查尿蛋白为阳性,2022年2月8日患者再次至我院复诊,完善24 h尿蛋白定量3 319.00 mg/24 h,24 h微量白蛋白定量2 813.00 mg/24 h,拟“蛋白尿查因”收入院。个人史无特殊。否认家族性肾脏病病史。入院查体未见特殊异常。入院后2022年2月10日查血常规:红细胞计数4.55×1012/L,血红蛋白浓度 127.00 g/L;肾功:肾小球滤过率评估 123.42 m1/分/1.73 m2,肌酐49.00 umol/L;血脂:总胆固醇 4.43 mmol/L,甘油三酯 5.41 mmol/L,低密度脂蛋白 1.25 mmol/L尿常规:红细胞计数 72.60/mL;抗磷脂酶A2抗体、抗核抗体、抗角蛋白抗体、抗环瓜氨酸抗体、抗肾小球基底膜抗体均为阴性,尿本周蛋白电泳阴性,类风湿因子、血管炎五项未见异常。2022-02-12查24 h尿蛋白定量3846.56 mg/24 h,24 h尿微量白蛋白定量3 234.56 mg/24 h。
1.1 肾脏穿刺活检结果
镜下检测到2个肾小球。肾小球基底膜无明显增厚,足突广泛融合,个别系膜区可见少量低密度电子致密物沉积,肾小球毛细血管襻开放,部分扩张,腔内见大量类脂空泡的蛋白物质。见图1。

图1 肾活检病理报告
1.2 组织学检查
送检肾穿刺组织可见21个肾小球,肾小球体积明显增大,毛细血管襻高度扩张,腔内充满淡染的、空泡状血栓样物质,系膜细胞和基质增生,局部可见系膜溶解,壁层上皮细胞无明显增生,未见新月体形成。见图2。
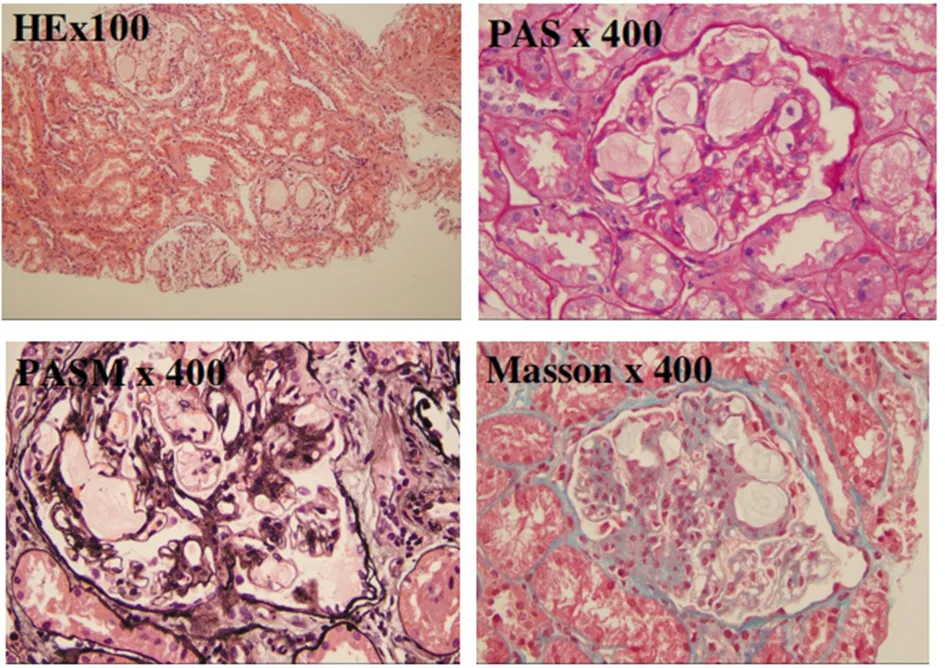
图2 常规肾脏病理检测(荧光7+特染3)
1.3 病理诊断
综合光镜、免疫荧光及电镜检查:高度疑为脂蛋白肾小球病。
免疫荧光:APOB:阴性;APOE:+,油红O:少数肾小管阳性。见图3。

注:A:ApoB;B:ApoE
1.4 基因检测
APOE基因存在c.394C>T(p.Arg132Cys)变异,见图 4。该变异为所致疾病临床表征与受检者临床表型及遗传模式相符,且变异评级为临床意义未明的变异。基因结果支持脂蛋白肾小球病的诊断。

图4 肾脏相关遗传病多基因测序
1.5 治疗及转归
结合患者病理结果及基因检测结果,脂蛋白肾病诊断明确,治疗上予肾炎康复片、替米沙坦片、百令胶囊护肾、降尿蛋白,非诺贝特胶囊降血脂,碳酸氢钠片碱化尿液对症治疗后患者临床症状及检验指标较入院前好转后出院。经治相关检验结果见图5。

注:A:尿蛋白折线;B:24 h尿蛋白折线图;C:血肌酐折线图;D:尿红细胞折线图
2 讨 论
脂蛋白肾病(LPG)的发现可追溯到30余年前,1989年日本学者Saito等[2]报道了一例前所未有的病例,其临床特征表现为难治性肾病综合征,组织学特征为肾小球内脂质样物质积聚,且无合并其他器官受累,清楚地将该病与其他遗传性脂代谢紊乱相关疾病区分开来。由于其免疫组织化显示肾小球沉积中存在脂蛋白,而该病的特征是血清脂蛋白水平异常,类似于Ⅲ型高脂血症的模式,因此这种疾病被命名为脂蛋白肾病。LPG人群发病率很低,有学者在中国知网、万方、维普、PubMed和Web of Science等数据库中检索了从数据库创建之年到2022年的LPG案例的文献,总共仅274例病例[1],其中这些病例以分布在东亚地区为主。虽然最初认为这种疾病仅限于东亚,以中国和日本多见,但已有几例高加索血统的病例报道[3]。
LPG是一种以组织病理学表现为肾小球毛细血管管腔扩张并在其内存在大量嗜酸性脂蛋白血栓,免疫荧光油红O染色呈阳性为特征的罕见的肾小球疾病。其发病机制尚未明确,目前主要认为由ApoE[2,4]的变异导致脂代谢异常引起。ApoE是一种由299个氨基酸组成的蛋白质,分子量约为39 kD。是人类脂蛋白(主要是富含甘油三酯的脂蛋白、其残基 和高密度脂蛋白)的组成部分,其最重要的生物学功能是作为一种配体[5],可促进胆固醇的转运和代谢,从而调节血液中血脂的水平,降低动脉粥样硬化性心血管疾病的发生风险[7]。ApoE基因的多态性可以影响ApoE浓度及各类脂蛋白的再分布,并参与慢性肾脏病的发生发展[8]。其中,ApoE基因突变引起结构和功能异常参与了ApoE2纯合子肾小球病和LPG等肾小球疾病[9],我们将这类疾病统称为ApoE 相关性肾小球病,临床主要表现为蛋白尿、肾功能不全及脂蛋白代谢异常、血清ApoE浓度显著增加等。而在LPG患者中,ApoE突变多发生在低密度脂蛋白(low density lipoprotein,LDL)受体结合位点,涉及LPG患者的140~150个氨基酸,ApoE突变后与LDL受体亲合力明显降低,此时ApoE多游离于血浆中,故LPG患者血中甘油三酯和ApoE升高[10],类似Ⅲ型高脂血症,但病变大多数仅限于肾脏。
目前发现的LPG 相关 apoE 基因突变共有10余种,呈常染色体显性遗传,在中国发现的LPG相关基因突变主要包括ApoE Kyoto、ApoE Tokyo/Maebashi、ApoE Guangzhou和ApoE Chengdu[11]。Oikawa 等[12]对ApoE 基因进行分析,发现ApoE 基因突变与 LPG 的发生有关,大多数是与 LDL 受体结合位点相近的杂合ApoE 基因错义突变,最常见的是ApoE-Sendai。然而,陈珊等学者[13]对5例正常对照组和17例脂蛋白肾病患者的ApoE基因全长经PCR扩增后进行测序分析,结果发现与正常对照组相比,17例脂蛋白肾病患者均并未存在ApoE基因突变,提示ApoE基因突变可能不是导致LPG的唯一因素[14]。
LPG有着患者年龄分布广、男性患者比例大于女性患者、地域分布的特点[15],其中日本和中国病例多见。LPG主要累及肾脏,以肾小球受累为主。几乎所有患者均以不同程度的蛋白尿为首发症状,并可伴发肾病综合征的体征,包括水肿、高血压等;少数患者也表现为轻微蛋白尿和镜下血尿。以本病例为例,患者以“排泡沫尿5月”为主诉入院,入院查24h尿蛋白定量升高,尿常规见红细胞,且肾活检病理也符合LPG病理特征。血脂异常并不是LPG的特征性改变,其血脂改变主要表现为血清Apo E 增高,但很少达正常值2倍以上[16],也可伴有类似于Ⅲ型高脂血症的症状如血浆胆固醇、甘油三酯、VLDL 升高,但III型高血脂血症引起的结节状或掌纹状黄瘤并未在LPG患者群体中观察到[5]。由于对该病认识时间较短,约50%的患者可在20余年内进展至终末期肾脏病[17],且进展至终末期肾病时患者血脂异常会更为明显[18]。LPG患者常见合并高血压,但合并恶性高血压很罕见,国内既往有报道1例LPG 患者合并血栓性微小血管病变(Thrombotic micro-angiopathy,TMA)导致恶性高血压的病例[19]值得关注,提示在LPG的诊疗中需注意有无小血管内血栓形成。目前未有关于LPG累及其他系统或合并动脉硬化和肝功能异常的报道,但多数LPG患者可出现不同程度贫血,且贫血与肾功能、小管间质病变无相关性[16]。
目前,LGP的诊断主要依靠其特征性的肾穿刺活检病理组织结果[20-21]。汪之玉等[16]学者汇总7例LPG患者的肾穿刺活检病理结果,其中肾小球体积增大为100%,肾小球分叶100%,襻扩张(>50%襻)为100%,襻内栓子(>50%襻)为100%,小管萎缩为50%,间质纤维化为50%;免疫荧光apoA为85.7%,apoB、apoE为100%;超微结果发现襻内血栓(>50%襻)为100%,系膜插入为57.1%,基膜增厚为100%。典型的LFG肾穿刺活检结果如下:光镜下可见肾小球体积明显增大,襻高度扩张,襻内见层状改变的大小不同、多少不等的“栓子”。多见轻至中度系膜增生变厚、系膜溶解,部分病例可见肾小球系膜基质向肾小球基底膜插入而形成的典型的“双轨征”。LPG的肾间质和肾血管多无明显的特征性病变。但随着疾病的进展,可进一步出现肾小球局灶甚至全肾小球硬化、肾小管萎缩及肾间质纤维化。肾小球毛细血管管腔内可见经 PAS、苏丹Ⅲ及 Masson 三色染色均呈弱阳性、经油红O染色呈强阳性的脂滴。免疫荧光检测见血栓样蛋白中含有大量脂类物,其中ApoE 和 ApoB 油红O染色阳性,普通染色较淡。个别可见IgM 或IgA少量沉积,多无免疫球蛋白、纤维蛋白原、补体沉积。电镜可见大小不等、电子密度不一的呈指纹样、簇状或层状排列在高度扩张的毛细血管管腔内的电子颗粒,免疫电镜证实沉积物中有被ApoE 和脂蛋白包绕的脂质。在LPG晚期,毛细血管管腔内脂蛋白栓子样物质减少,系膜细胞及基质高度增生伴节段性系膜插入及硬化,增生的系膜逐渐取代脂蛋白栓子,因此不易诊断脂蛋白肾病为原发病变,反而会被误诊为局灶节段性肾小球硬化症、增生硬化性肾炎及膜增生性肾炎。同时,ApoE 和 ApoB 染色的免疫组化技术是该病诊断与鉴别诊断的关键。
目前,LPG尚无明确且有效的治疗方法,既往研究发现,糖皮质激素、免疫抑制剂、抗凝疗法对 LPG 基本无效[6,22],目前主要的治疗方法仅局限于针对大量蛋白尿、水钠潴留、高血脂症的对症治疗[1]。但国内外先后有不同类别降脂药、肾移植、血液净化及免疫吸附方法治疗 LPG 达到缓解的个案报道。Cheung等[23]学者报道了一例罕见的LPG合并纤维性肾小球肾炎并发展为终末期肾脏病的中国年轻患者,该患者接受了已故肾移植,在移植后20年的随访中被诊断为LPG复发。该病例是文献中肾移植后随访时间最长的病例,表明LPG患者的肾移植预后仍然良好。肾移植应该仍然是LPG引起的ESKD患者的一种治疗选择。2013年,范文静等[22]通过30例随访时间大于2年的LPG患者,其中15例患者用非诺贝特治疗,15例用安慰剂设为对照组,治疗加随访24个月后发现非诺贝特可降低脂蛋白肾病患者尿蛋白及血 ApoE 水平,延缓肾功能不全的进展。2015 年 Li 等[24]报道了1例临床表现为大量蛋白尿的 LPG 患者,经双重血浆置换治疗3次后,尿蛋白转阴的案例。2008年,Xin等[25]学者给与13 名肾活检证实为 LPG 的患者葡萄球菌蛋白A免疫吸附治疗,单次免疫吸附治疗后LPG患者蛋白尿及血清apoE显著下降,12名患者重复肾活检显示肾小球内脂蛋白血栓几乎消失。6名患者参加了长期结果的调查,蛋白尿在12个月内恢复到基线水平,结果表明蛋白A免疫吸附可能是解决LPG患者肾小球内血栓和改善肾病综合征的有效方法,但该组病例随访时间较短,需要进一步的研究来确定免疫吸附对 LPG 患者的长期影响。目前关于LPG患者接受中医药治疗的报道尚少,2019年,高颖颖[26]等通过中医药固肾健脾益气、利湿、活血通络辅以西药常规降脂治疗1例LPG患者23天,治疗后患者蛋白尿明显改善,出院后半年规律随诊,24 h尿蛋白定量波动在2200 mg/24 h,甘油三酯波动在2.10 mmol/L,疗效稳定。但该案例病例数量较少且追踪时间段短,需要进一步的研究来证实中西医结合治疗LPG的可行性。还有学者发现,抗惊厥类药物托吡酯可以减轻受试者体重,在一定程度上降低血脂和血糖,Manzini等[27]通过动物实验,发现托吡酯可以保护APOE缺陷小鼠免受肾脏损害,为LPG的治疗提供了新的思路。
综上所述,LPG 是人体ApoE基因突变后导致的一种以组织病理学表现为肾小球毛细血管管腔扩张并在其内存在大量嗜酸性脂蛋白血栓,免疫荧光油红O染色呈阳性为特征的罕见的肾小球疾病。由于LPG 缺乏特征性的临床表现,多表现为肾病综合征,少数患者出现血尿、镜下血尿、血脂升高,因此,不能仅仅通过临床表现诊断 LPG,目前肾脏活组织穿刺病理检查及基因检测是诊断 LPG 最重要的手段。关于LPG可能存在的发病机制及有效治疗方案亟待被发现。
