基于Cu-Ce复合氧化物的低浓度CH4催化燃烧性能及微观机理
2022-03-21张晨航豆宝娟滕子豪吴亮锴郝庆兰
张晨航,豆宝娟,滕子豪,吴亮锴,郝庆兰,宾 峰
(1.天津科技大学 海洋与环境学院,天津 300222; 2.中国科学院力学研究所 高温气体动力学国家重点实验室,北京 100190)
0 引 言
我国每年通过乏风瓦斯向环境中排放大量的甲烷气体,作为重要的温室气体,甲烷对全球增温的能力是CO2的21倍[1]。乏风瓦斯热值比较高(甲烷的热值为35.9 MJ/m3),若能够将其合理利用,具有环境保护和能源利用的双重价值。目前,催化燃烧技术[2]被视为乏风瓦斯的潜在处理技术之一。甲烷,化学式CH4,是最简单的烃,由1个碳和4个氢原子通过sp3杂化的方式组成,因此甲烷分子的结构为正四面体结构,4个键的键长相同键角相等。因其具有4个稳定的C—H键,键能达到413 kJ/mol,甲烷难以被一般的催化剂活化[3],故设计高性能的催化剂使其适用于甲烷的催化燃烧就成了解决乏风瓦斯高效燃烧问题的关键。
国内外许多学者针对甲烷催化氧化开展了大量试验研究[4-6],典型的催化剂为负载型贵金属和过渡金属氧化物。贵金属催化剂尽管具有很高的活性,尤其是Pd和Pt,然而在较高温度下易烧结和升华,容易导致活性组分流失。与贵金属相比,负载型过渡金属氧化物虽然活性比较低,但是在苛刻的环境条件下更具有耐热性、抗毒性强等特点,相比之下具有更高成本效益。
在非贵金属类催化剂方面上,铜-铈催化剂因具有较高的活性和耐久性,被视为贵金属催化剂的替代品[7]。其中,Cu基催化剂本身也具有优良的催化性能和耐久性,在500~700 ℃时能够实现乏风瓦斯的氧化,但Cu基催化剂的塔曼温度较低[8],在甲烷催化燃烧中单独使用,易因高温导致烧结而失活,若将Cu负载在另一种过渡金属氧化物表面,即可获得显著的耐烧结性能,且载体上的Cu基氧化物具有较高的分散度,进而降低了团聚现象[9]。除了抑制烧结及团聚现象以外,双金属、多金属的协同增效作用有利于催化活性的提高。CHOUDHARY等[10]研究发现Cr、Co、Mn等过渡金属氧化物掺杂在Cu基催化剂中使甲烷的燃烧活性显著提高,原因是过渡金属氧化物的掺杂致使催化剂内部形成一定的晶格缺陷,以提高氧的迁移效率。CH4催化反应的主要活性位是CuOx。相关研究证实[11],高度分散的CuOx簇通常被确定为CuO-CeO2的活性成分,而CeO2作为助剂,其特有的萤石型结构能够有效促进Cu物种的分散,并且Ce会通过Ce4+和Ce3+的变换,使得CeO2具有优良的储放氧性能。KANG等[12]研究发现CO氧化反应主要发生于CuO-CeO2表面分散的CuOx和CeO2相邻的氧空位交界处。但是目前对CuO、CeO2和CuO-CeO2在CH4催化燃烧反应过程中活性位确定的相关研究较少,同时由于CH4分子的结构稳定性,一般催化剂表面的活性氧通常很难与目标气体结合。Cu-Ce催化剂以其高效的协同特性与活化性能被应用于结构稳定气体的催化燃烧。现有的研究针对Cu、Ce及Cu-Ce活性位点对CH4催化燃烧反应的作用机制尚不明晰。为此,笔者采用溶胶凝胶法制备CuO、CeO2和CuO-CeO2三种催化剂,首先采用XRD、XPS、H2-TPR和O2-TPD等方法考察其物理化学特性,再利用原位红外光谱和分压反应动力学方法研究反应物的化学吸附和反应途径。该研究成果将为催化燃烧甲烷的催化剂活性位点和微观反应机理提供新的见解,为乏风瓦斯大气污染治理及余热高效利用提供借鉴。
1 试验方法
1.1 CuO/CeO2/CuO-CeO2的制备
Cu(NO3)2·3H2O和Ce(NO3)3·6H2O试剂以物质的量比1∶1混合,完全溶解在乙醇溶液中(80 ℃、140 mL),再快速加入草酸溶液(0.24 mol/L、造孔剂),恒温搅拌溶液使乙醇蒸发,直至形成凝胶。凝胶在室温下老化48 h后,在105 ℃下干燥12 h,然后在550 ℃下煅烧2 h,所得催化剂记为CuO-CeO2。为了比较铜氧化物和铈氧化物对CH4氧化性能的影响,采用相同的方法分别制备CuO和CeO2催化剂。
1.2 样品表征
催化剂的晶体结构由XD-3型全自动衍射仪测得(XRD),扫描方式和速度分别是2θ/θ连续扫描和0.02 ℃/min。采用共聚焦显微拉曼光谱仪(HORIBA LabRAM HR Evolution)在488 nm的激光波长下测定催化剂的氧空位浓度(光谱范围:10~9 000 cm-1,行程范围:112 mm×76 mm)。使用Kratos Axis Ultra DLD型X射线光电子能谱仪光谱仪进行了XPS分析。在测试前,将分析室腔体内的真空度抽到5.0×10-7Pa 。在TP5080B化学吸附仪上开展程序升温还原(H2-TPR)分析:20 mg催化剂置于石英反应管中,在5% H2/Ar中以10 ℃/min的程序升温速率升至500 ℃;改通100% O2后,在O2中吸附30 min,然后暴露于N2中升温至300 ℃再降到室温,改通O230 min,用N2吹扫20 min用来吹掉表面物理吸附的O2,再以10 ℃/min程序升温速率升温至980 ℃,全程使用热导池检测器进行跟踪检测。采用TP5080B化学吸附仪探究催化剂表面吸附或氧化的情况(O2-TPD)。使用在线气体分析仪(Maihak)来监测流出的CH4、O2和CO2(CH4体积分数为0%~30%,精度为±2%FS;O2体积分数为0~40%,精度为±3%FS;CO2体积分数为0~10%,精度为±3%FS)。在FOLI10-R仪器上开展原位红外光谱研究,采用磁驱动透射式原位池,用50 mL/min的N2升温到300 ℃进行预处理,降到室温后,采用总流量50 mL/min的10%CH4+90%Air进行背景光谱采集,其中约3 mg催化剂和溴化钾被压入支撑架中开展CH4氧化反应探究。
1.3 催化剂活性评价
CH4催化燃烧的活性评价在固定床反应器上进行,气体流量用质量流量计控制,总流量为200 mL/min,反应气的体积组成为1% CH4,78% N2和21% O2。催化剂质量0.2 g,填充于内径为3 mm的石英玻璃反应管中,试验采用程序升温的方法,温度控制在100~550 ℃,升温速率为5 ℃/min。反应前后的气体组分及浓度由北京麦哈克红外气体分析仪(QGS-08)在线测定。
2 结果与讨论
2.1 XRD分析
CuO、CeO2和CuO-CeO2的XRD衍射谱图如图1所示。从图1中均可观察到3种催化剂均以典型的CeO2立方萤石结构存在,衍射峰位于28.5°,33.1°,47.5°和56.3°,分别对应(111)、(200)、(220)和(311)晶面[13]。另外,图1中35.6°和38.8°归属CuO的衍射峰,对于CuO和CuO-CeO2催化剂,CuO-CeO2催化剂中该峰强度与CuO相比明显减弱,并结合其萤石结构峰型宽化现象,表明CuO-CeO2催化剂形成CuCeOx固溶体或以分散性良好的CuO团簇形式存在,并锚定于CeO2表面[12]。

图1 3种催化剂的XRD衍射图Fig.1 XRD diffraction map of CuO, CeO2,and CuO-CeO2 catalysts
2.2 XPS分析
催化剂表面元素组成和化学状态由XPS测定。Cu 2p在催化剂上的XPS谱图如图2所示,Cu 2p3/2峰具有不对称性,原始曲线可以分峰拟合为933.2 eV和934.8 eV,分别对应于Cu+和Cu2+,这表明在CuO和CuO-CeO2催化剂中存在着Cu+/Cu2+氧化还原对。此外,941~944 eV处出现的卫星峰也证明了Cu2+的存在。

图2 Cu 2p在催化剂上的XPS谱图Fig.2 Cu 2p XPS spectrum in the catalysts
Ce 3d能谱由8个峰位组成(图3),将其分解成4对自旋轨道双峰(用u、v标记):其中u、u2和u3峰对应于Ce4+的3d3/2水平,而标记为v、v2和v3的峰分配给Ce4+的3d5/2;882.6和900.8 eV处分别归属为Ce 3d5/2和Ce 3d3/2。CuO-CeO2催化剂4个强度比较高的882.6、900.8、898.3、917.0 eV峰归属于Ce4+物种中不同的Ce 4f电子排布,2个比较弱的峰(889.3、907.3 eV)代表Ce3+中可能得电子情况下2种排布的一种。Ce4+是正六面体稳定结构,可以增加活性物种CuO的分散状态,同时Ce3+不是正六面体结构且氧空位数量多,所以增强Ce3+对O的吸附能力,Ce在三价和四价的转换显著提升催化剂储氧放氧能力因此能够提高催化剂活性。根据表1列出的铜和铈物种的表面组成,CuO-CeO2的Cu+/Cu2+和Ce3+/Ce4+比分别高于块状CuO和CeO2,表明这些Cu+物种产生于Cu+-[Ov]-Ce3+界面([Ov]=表面氧空位)通过Ce4+还原形成了Ce3+。
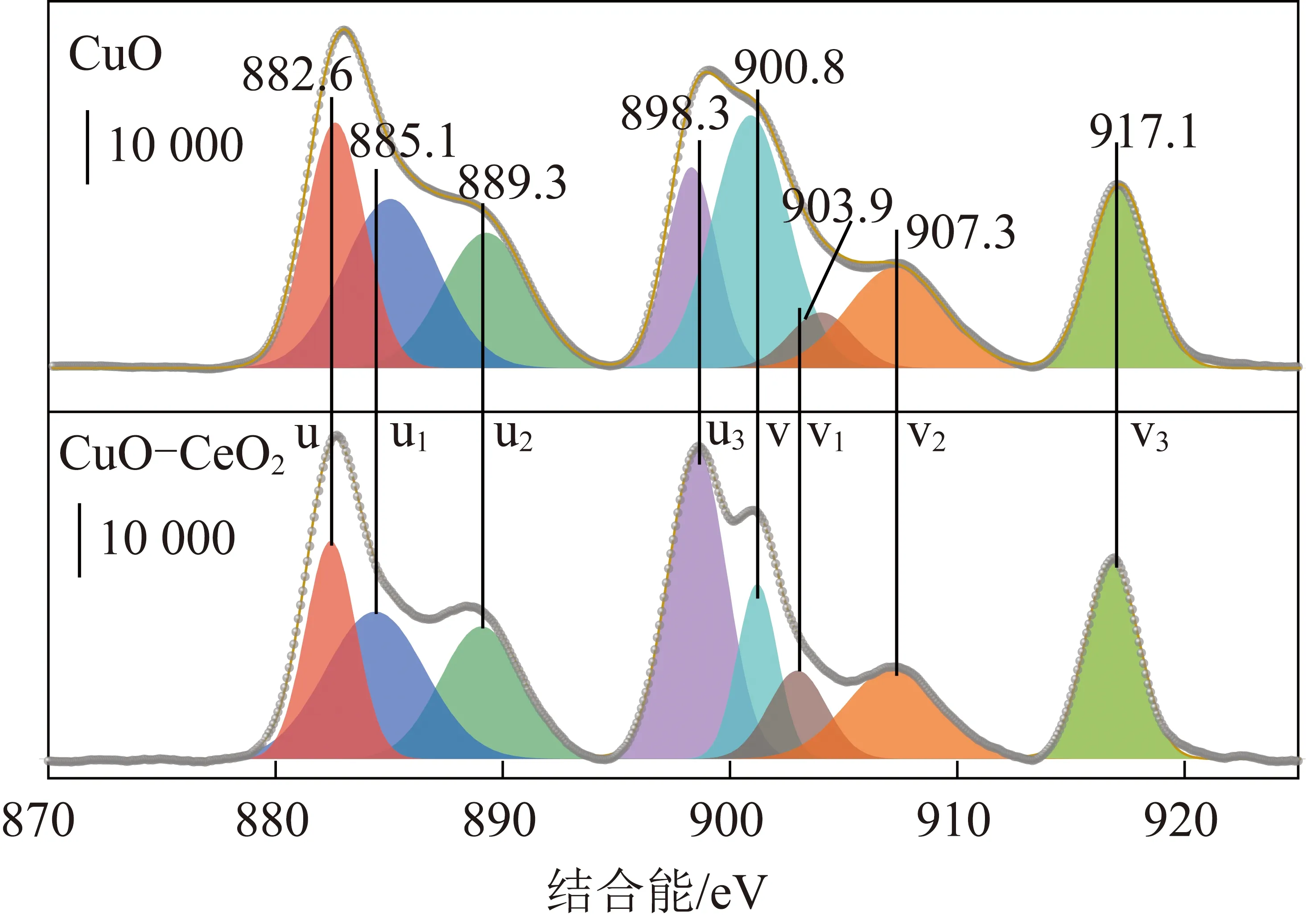
图3 催化剂上的Ce 3d XPS谱图Fig.3 Ce 3d XPS spectrum in the catalysts

表1 催化剂的表面组成
2.3 H2-TPR试验结果分析
采用H2-TPR测试催化剂的氧化还原能力,如图4所示,CuO的还原峰由216 ℃的γ峰和184 ℃的β峰组成,分别归因于分散的CuOx团簇和结晶CuO中铜物种的还原[12]。而CuO-CeO2催化剂在153 ℃(α峰)、186 ℃(β峰)和202 ℃(γ峰)有3个重叠的还原峰,其中α峰归因于CuCeOx固溶体的还原。CuO-CeO2中α峰的温度较低就证实了与氧结合的能力强于CuO,而CuO和CeO2之间的协同效应导致CuO-CeO2比纯CeO2更低的还原温度(α峰的存在)。3种催化剂的氢气消耗量见表2。由表2可知,CuO催化剂的H2消耗量最大,其原因是铜物种比铈物种更易还原,且该催化剂的铜含量最高,CuO-CeO2的H2消耗量次之,CeO2耗氢量最少。需要指出的是,催化剂的氧化还原性能主要取决于其较低的还原温度,所以3种催化剂的氧化还原能力排序为CuO-CeO2>CuO>CeO2。

图4 3种催化剂的H2-TPR曲线Fig.4 H2-TPR curves of CuO, CeO2, CuO-CeO2 catalysts

表2 3种催化剂的H2消耗量
2.4 O2-TPD试验结果分析
采用O2-TPD方法考察催化剂中氧物种的迁移率,如图5所示。CuO和CuO-CeO2催化剂的O2-TPD曲线分别在300 ℃和800 ℃左右出现低温峰和高温峰。CuO、CeO2和CuO-CeO2三种催化剂的低温峰主要由于表面物理吸附氧脱附引起,而CuO和CuO-CeO2的高温峰来源于催化剂中的体相晶格氧脱附。在高温段,CuO催化剂在820 ℃的脱附峰对应从Cu离子逃逸的晶格氧,CeO2催化剂良好的热稳定性致使体系中无晶格氧脱附,XPS结果也证实,纯CuO是一种非化学计量比氧化物,晶格氧脱附后所形成的金属离子缺陷比CeO2更容易吸附氧。纯CeO2上的氧空位对氧吸附的贡献十分有限。然而,铜和铈之间的协同效应导致氧的解吸峰向低温(790 ℃)移动。尽管CuO-CeO2的脱附峰面积小于CuO催化剂的峰面积,但脱附温度下降,表明CuO-CeO2催化剂有更强的氧流动性,加速氧从内部到表面的迁移,有利于高温下的氧化反应,从而提高甲烷催化燃烧的催化活性。

图5 CuO、CeO2、CuO-CeO2催化剂的O2-TPD图Fig.5 O2-TPD diagram of CuO, CeO2 and CuO-CeO2 catalysts
2.5 催化剂活性评价
CH4催化燃烧的活性评价在自行建立的固定床反应器上进行,反应气总流量控制为200 mL/min(1% CH4、78% N2和21% O2),所得到的CH4催化燃烧活性曲线如图6所示。3种催化剂的活性顺序为CuO-CeO2>CuO>CeO2。CeO2的T10(转化率为10%所对应的温度)为438 ℃,T90(转化率为90%所对应的温度)为530 ℃,效果最差;效果较好的CuO的T10和T90分别为348 ℃和483 ℃,而效果最好的CuO-CeO2催化剂T10为323 ℃,T90为444 ℃,结合XRD和XPS分析结果,研究表明CuO与CeO2相互掺杂形成一定的晶格缺陷,这些缺陷提高了氧的迁移速率,从而促进CuO-CeO2催化活性明显提升。据文献[14-16]报道,Cu系催化剂对甲烷的转化率与负载量呈正比关系,指出负载量过高会导致金属分散度降低和表面活性位堆积。然而本研究引入金属Ce不但带来更高的移动活性氧,也提高了Cu的分散性。故CuO-CeO2催化剂在催化氧化CH4的活性评价中表现出比CuO和CeO2更高的低温活性。

图6 CuO、CeO2、CuO-CeO2催化剂催化燃烧CH4的转化率曲线Fig.6 Conversion rate curves of CH4 combustion overthe CuO, CeO2 and CuO-CeO2 catalysts
2.6 分压反应动力学分析
为了探究CH4与O2对反应速率的影响规律,在催化剂活性评价台上进行分压反应动力学试验,反应过程中CH4的转化率控制在<10%。保持CH4或O2压力不变,N2作为平衡气,总流量200 mL/min,基于CuO-CeO2催化剂确定CH4催化燃烧反应速率方程和O2对反应速率的影响规律,其动力学试验结果见表3。

表3 CuO-CeO2催化剂的动力学试验结果
笔者采用Polymath 6.10建立了基于质量作用定律的动力学经典模型:
r(CO)=kPa(CH4)Pb(O2),
(1)
其中,r(CO)为CO转化率;k为反应速率常数,s-1;P(CH4)和P(O2)为CH4分压和O2分压;a和b为CH4和O2的反应级数。经过模型计算,CuO-CeO2的动力学模型为:r(CO)=1.33P(CH4)1.029P(O2)0.948,曲线拟合的相关系数大于0.999。因此,CuO-CeO2催化剂CH4(1.029)和O2(0.948)的反应级数相差不大(图7),但CH4的反应级数大于O2,反应遵循L-H机理,即吸附的CH4与吸附的O2发生反应。

图7 CuO-CeO2催化剂的CH4催化燃烧反应速率Fig.7 CH4 reaction rate of the CuO-CeO2 catalyst
2.7 In-situ DRIFT结果分析
为进一步分析CH4催化燃烧过程的吸附中间产物,采用原位红外光谱仪探究了CuO-CeO2催化剂上CH4催化燃烧反应过程,结果如图8所示。
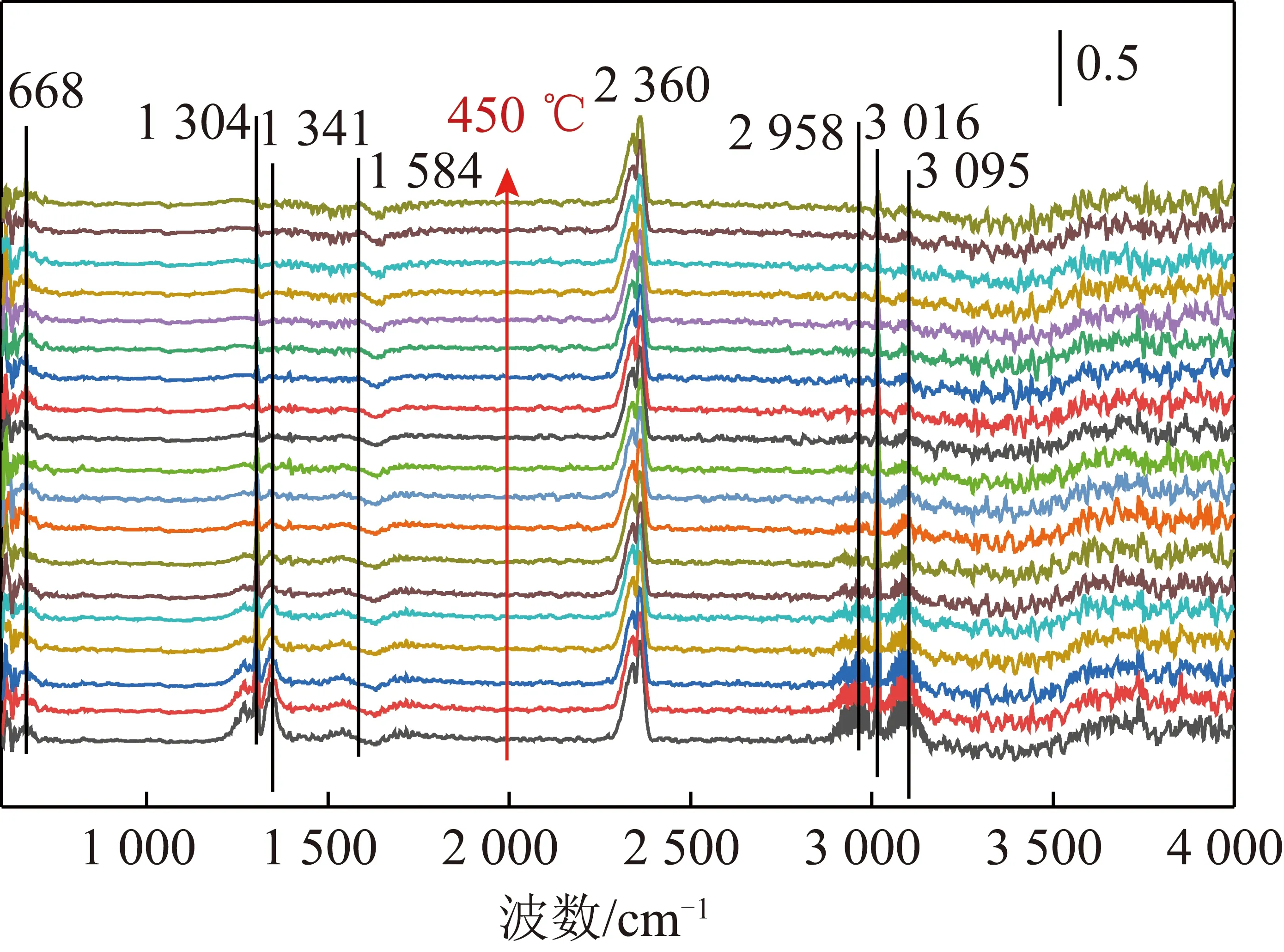
图8 CuO-CeO2上CH4催化燃烧原位红外光谱分析Fig.8 In-situ infrared spectroscopy analysis for CH4 catalyticcombustion over CuO-CeO2
由图8可知,50 ℃时,波数为1 304、1 341及1 584 cm-1处的特征峰主要归属于CH4与O2吸附在CuO-CeO2催化剂CuCeOx固溶体上形成碳酸盐(RCO3)物种[17-20]。2 958、3 016 及3 095 cm-1处为CH4化学吸附在催化剂上的CuCeOx固溶体及分散性CuOx的Cu位点上,形成了甲氧基物种(Cu—OCH3)与甲酸盐物种(Cu—OOCH)[21]。2 360 cm-1处的特征峰归属于CO2的产生峰。随着反应温度的升高,甲氧基与甲酸盐物种的不饱和C—H键的强度急剧降低,与吸附氧反应生成了CO2;同时碳酸盐分解也产生了CO2。由以上结果可知,CuCeOx固溶体及分散性CuOx是反应的主要活性位点,吸附的甲烷与吸附的氧生成中间物种(甲氧基、甲酸盐及碳酸盐),进而分解产生CO2和H2O,反应遵循L-H机理,结果与反应动力学结果一致。
3 结 论
1)使用溶胶凝胶法制备了3种催化剂(CuO/CeO2/CuO-CeO2),并通过催化剂活性评价,发现CuO-CeO2催化剂在甲烷催化燃烧表现出了最佳活性,CH4开始转化温度为246 ℃,完全转化温度461 ℃。
2) XPS、XRD、H2-TPR、O2-TPD结果表明过渡金属氧化物CuO的掺杂,引入了一定量的晶格缺陷,提高了氧的迁移速率。Ce3+/Ce4+和Cu2+/Cu+电子对均参与CH4的催化循环(Cu++Ce4+←→Cu2++Ce3+),活性氧的主要来源是CuO。同时Ce3+和Ce4+的转换也提高催化剂的储氧放氧能力,Ce的引入提高了Cu在催化剂表面的分散性,并且形成了CuCeOx固溶体。CuO-CeO2催化剂上分散性CuOx与CuCeOx固溶体是主要的反应活性位,促进活性的提高。
3) 提出了甲烷在CuO-CeO2催化剂上可能存在的反应机理。通过分压动力学试验及模型计算表明,在催化燃烧过程中,CuO作为CH4和与O2的吸附活性位,在催化剂表面的反应级数相差不大且接近1(CH4:1.029,O2:0.948),即吸附的CH4与吸附的O2发生反应,反应遵循L-H机理。同时原位红外试验结果表明,吸附的甲烷与吸附的氧生成中间物种甲氧基、甲酸盐及碳酸盐。甲氧基与甲酸盐物种的不饱和C—H键与吸附氧反应以及碳酸盐的分解产生CO2和H2O,反应遵循L-H机理,2者进行了相互验证。
