聚苯胺在芳纶Ⅲ表面的原位聚合及产物吸波性能研究
2022-03-19胡书春张瑶涵李旭勤任亚琦
王 哲,胡书春,张瑶涵,彭 涛,李旭勤,任亚琦
(1.西南交通大学 材料科学与工程学院,四川 成都 610031;2.成都工业学院 材料与环境工程学院,四川 成都 611730; 3.西南交通大学 材料先进技术教育部重点实验室,四川 成都 610031;4. 中蓝晨光化工研究设计院有限公司,四川 成都 610041)
吸波材料是一种以吸收为主,反射、散射和透射都很小的一类功能材料,在军工和民用领域均有广泛的应用基础[1-3]。吸波材料的研制及其在武器装备隐身领域的应用,已成为世界众多国家重点发展的高新及国防技术[4-6]。目前,在军用飞机、弹道导弹等武器装备的隐身设计领域,除了对吸波材料强吸收效能的要求之外,也对其轻质性和力学性能提出了更高的要求。近年来,在轻质吸波材料领域,聚苯胺(PANi)凭借其密度低、电磁损耗能力优异、导电性高和易于制备等优势,获得了国内外学者的广泛关注[7-8]。颜海燕等[9]采用化学氧化法制备了分别以盐酸(HCl)、硫酸(H2SO4)、十二烷基苯磺酸(DBSA)、甲基苯磺酸(TSA)和磺基水杨酸(SSA)掺杂的PANi产物,发现在9.3 GHz时,PANi-HCl和PANi-DBSA吸波性能最为优异,其反射损耗(RL)值分别为-4.94 dB和-5.12 dB。H.MIZOBUCHI[10]用氨基萘磺酸掺杂合成PANi,制得了粒径在8~12 nm的铁磁性导电PANi产物,发现该产物在1~18 GHz频段兼具电损耗和磁损耗能力。
虽然PANi拥有轻质性和良好的电磁波损耗能力等优点,但也存在力学性能不够优异的短板,限制了其在结构功能一体化的武器装备隐身设计领域的应用。基于此,国内外研究团队开展了在纤维材料表面合成PANi的研究工作,以提升PANi类吸波材料的机械力学性能。P.SAINI等[11]分别以聚丙烯酸和聚苯乙烯磺酸掺杂的PANi在棉纤维表面负载,发现改性PANi产物表现出较好电磁屏蔽效能,二者的最大屏蔽效能值分别为-5.2 dB和-5.7 dB,对应的电磁波频率(f)均为12.5 GHz。相对于棉纤维,芳纶III(F3)具有更为优异的综合性能,其拉伸强度可达4.5 GPa,弹性模量可达130 GPa[12]。基于F3优异的性能,如果在F3表面实现导电高分子的原位合成与沉积,极有可能获得兼具优异吸波性能和突出机械力学性能的结构功能一体化导电高分子类吸波材料。作者所在课题组前期已报道了聚吡咯在F3表面的原位负载及产物吸波性能研究工作[13],本研究将进一步开展PANi在F3表面的原位合成,并对合成产物的宏观性状、微观结构与形貌进行表征和分析研究,并在此基础上提出PANi在F3表面的原位合成过程与机理,最后对合成产物的电磁学和吸波性能进行深入研究。
1 实验
1.1 原料及试剂
F3:中蓝晨光化工研究设计院有限公司产;苯胺(AN)、对甲苯磺酸(PTSA)、HCl、过硫酸铵(APS)、无水乙醇:分析纯,成都科龙化工试剂厂产;石蜡:工业级,国药集团化学试剂有限公司产;蒸馏水:实验室自制。
1.2 PANi在F3表面的原位合成
用FLS-tpj-4型电剪刀将F3截切成5 mm左右的短纤维,并用适量无水乙醇和蒸馏水分别清洗截切后的F3,然后对其进行烘干处理(55 ℃,12 h);称取0.4 g烘干后的F3,将其加入到180 mL蒸馏水中,充分搅拌至F3分散均匀;向体系中先后加入1 g AN单体和浓度为0.35 mol/L的PTSA溶液6 mL,然后将混合液保持在20 ℃恒温水浴中,并缓慢机械搅拌40 min;接着向混合液中滴加20 mL预先配制的APS水溶液(浓度为0.07 mol/L),滴速控制在2 mL/min左右;滴加结束后,保持在20 ℃水浴和缓慢搅拌条件下反应5 h,然后对反应产物进行减压抽滤,并用蒸馏水和无水乙醇交替清洗至洗涤液无色和澄清透明;最后经烘干(55 ℃,12 h)处理,得到在F3表面原位合成有PTSA掺杂PANi的复合纤维产物,记为PTSA-PANi/F3。
保持以上实验过程和参数不变,仅将PTSA溶液变更为HCl溶液(6 mL,0.35 mol/L),制备出在F3表面原位合成有HCl掺杂PANi的复合纤维产物,记为HCl-PANi/F3。
1.3 分析与测试
宏观性状与微观形貌:采用日本尼康公司D5300型数码相机对F3和PTSA-PANi/F3、HCl-PANi/F3产物的外观性状进行数码照片拍摄,并进一步用美国FEI公司Quanta 200型扫描电子显微镜(SEM)观察F3和PTSA-PANi/F3、HCl-PANi/F3产物的微观形貌,工作电压20 kV。
红外光谱(FTIR):采用美国赛默飞世尔科技公司Nicolet 6700 型傅里叶红外光谱仪对F3和PTSA-PANi/F3、HCl-PANi/F3产物进行测试,KBr压片,波数为400~4 000 cm-1。
X射线衍射(XRD):采用荷兰帕纳科公司Philips X′Pert PRO型X射线衍射仪对F3和PTSA-PANi/F3、HCl-PANi/F3产物的物相与结晶结构进行分析。辐射光源为Cu-Kα,操作电压40 kV,操作电流30 mA,扫描角度(2θ)10°~70°。
电导率:将F3和PTSA-PANi/F3、HCl-PANi/F3产物分别制成厚度约为1 mm的圆片状试样,然后用广州四探针科技有限公司RTS-9型双电测四方探针测试仪测试试样的电导率。
复介电常数和复磁导率:将F3和PTSA-PANi/F3、HCl-PANi/F3产物分别与石蜡按质量比3∶7 混合后,然后放入同轴样品制备专用模具中,并在20 MPa载荷下保压2 min得到3种纤维样品的同轴试样(其外径为(7±0.02) mm,内径为(3.04±0.02) mm,厚度为4 mm左右)。利用制备得到的同轴试样,在中国电子科技集团公司AV3672C型微波一体化矢量网络分析仪上测试三种试样在8~12 GHz 频段的复介电常数和复磁导率,测试点数为51点。
2 结果与讨论
2.1 宏观性状与微观形貌
从图1可以看出,原料F3呈蓬松堆积状态、外观呈黄色,其改性产物PTSA-PANi/F3和HCl-PANi/F3的堆积状态较F3致密,外观分别呈墨绿色和灰黑色。

图1 试样的数码照片Fig.1 Digital photos of samples
这一方面是由于F3的表面原位负载PANi后提升了改性纤维之间的吸附力;另一方面是因为原位合成工艺实现了PANi在F3表面的原位生成及对F3骨架的致密包覆,所以两种改性产物的外观颜色呈现了PANi的颜色。由于本研究的PANi是通过化学氧化聚合途径制备的,其颜色受氧化程度影响,在氧化程度较低时可能呈现墨绿色,而在氧化程度较高时可能呈现灰黑色[14]。其中质子酸在AN合成反应中除起到掺杂作用外,还为氧化剂APS的分解和自由基的形成提供酸性环境[15],因此可以推断出质子酸的酸性强弱会影响PANi的氧化程度,所以在本实验条件下,HCl掺杂较PTSA掺杂会提升PANi的氧化程度,进而导致HCl-PANi/F3产物较PTSA-PANi/F3产物的颜色更深。
从图2可以看出,原料F3的表面光洁,而PTSA-PANi/F3和HCl-PANi/F3产物仍呈纤维状,但在其纤维表面均明显生成有致密的膜层物质,HCl-PANi/F3产物中的PANi膜层更为致密和均匀,而PTSA-PANi/F3产物则明显存在局部PANi膜层凸起的问题,导致其膜层连续性较差及在不同纤维区域的沉积厚度存在差异。

图2 试样的SEM照片Fig.2 SEM images of samples
2.2 FTIR分析
从图3可以看出,在原料F3的FTIR图谱中,3 295 cm-1处的吸收峰对应N—H伸缩振动,1 655 cm-1处的吸收峰对应酰胺基团中的C-O键的伸缩振动,1 597 cm-1和1 246 cm-1处的两个吸收峰分别对应苯并咪唑环中的C-N伸缩振动和N-H弯曲振动[16],1 510 cm-1、1 474 cm-1和1 408 cm-1处的三个吸收峰分别对应苯环上的C-C、C—N和C—C的伸缩振动峰[16-17],1 110 cm-1和1 016 cm-1两个吸收峰分别对应苯环上C—H和N—H的面内弯曲振动[18],956 cm-1处的吸收峰对应苯环上C—H的面外弯曲振动,827 cm-1到660 cm-1之间的多谱带对应苯环的多取代吸收峰。在PTSA-PANi/F3、HCl-PANi/F3产物的FTIR图谱中,并未出现明显区别于F3的新吸收峰。但是通过对比发现,3 295 cm-1处的N—H峰红移动到了3 435 cm-1处,这主要归因于在F3分子与PANi之间形成了分子间氢键[19]。1 306 cm-1处吸收峰强度的增大主要归因于F3的C—N伸缩振动与PANi中C—N伸缩振动峰的叠加[20]。1 484 cm-1处吸收峰峰形的拓宽是由于F3的C-C、C—N伸缩振动峰与PANi中苯环骨架的C-C伸缩振动峰合并所致[21]。1 110 cm-1处吸收峰的峰形拓宽是因为F3的C—H面内弯曲振动峰与PANi醌式N-Q-N吸收峰的合并所致[22]。与原料F3相比,在两种PANi/F3产物的FTIR图谱中,956 cm-1、1 110 cm-1和1 016 cm-1处的三个吸收峰强度减弱,表明PANi在F3表面的裹覆对苯环上的C—H和N—H的面内弯曲振动及C—H的面外弯曲振动都产生了限制,这主要归因于PANi在F3表面的原位合成过程中除了具有物理吸附作用以外,还与F3形成了分子间氢键,进而导致F3的苯环和PANi的醌环通过氢键连接起来,并进而对苯环上C—H和N—H的弯曲振动产生了更大的位阻。综合SEM和FTIR分析结果可知,在两种PANi/F3产物中,PANi原位聚合在了F3表面,两种物质之间除了物理吸附作用以外,还存在氢键的结合。
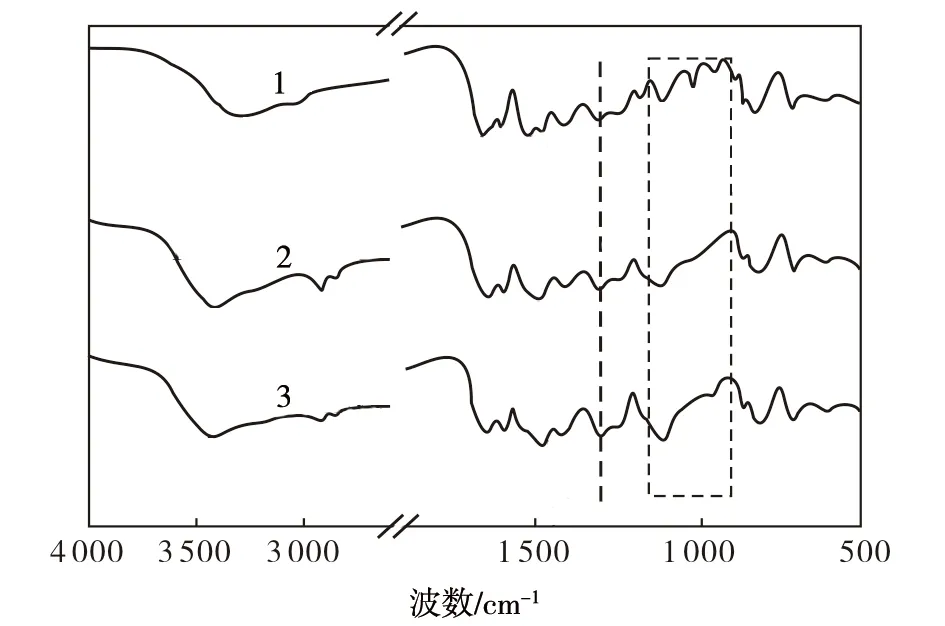
图3 试样的FTIR图谱Fig.3 FTIR spectra of samples1—F3;2—PTSA-PANi/F3;3—HCl-PANi/F3
2.3 XRD分析
从图4可以看出:原料F3在2θ为20.5°处有一个尖峰,对应于F3特征衍射峰(110)晶面,且此峰强度大,峰形窄,表明原料F3具有较高结晶度,采用Hinrichen方法[23]计算得到F3的结晶度为46%;对于HCl-PANi/F3、PTSA-PANi/F3均在2θ为19°附近存在一个肩形突出,峰形宽但强度较低,这主要归因于在F3表面原位合成的PANi呈无定型结构,在2θ为26°处产生无定型结构的X-ray衍射峰[24],并与F3的(110)晶面衍射峰合并。由于本研究中的PANi主要通过AN单体在F3表面的化学氧化聚合而生成,并通过物理吸附和氢键作用而沉积和包覆于F3骨架表面,因此可以推断这种原位聚合和沉积过程不会对F3的结晶结构产生明显影响,这有益于依托F3的高强度和高模量来赋予HCl-PANi/F3和PTSA-PANi/F3产物较为优异的机械力学性能。
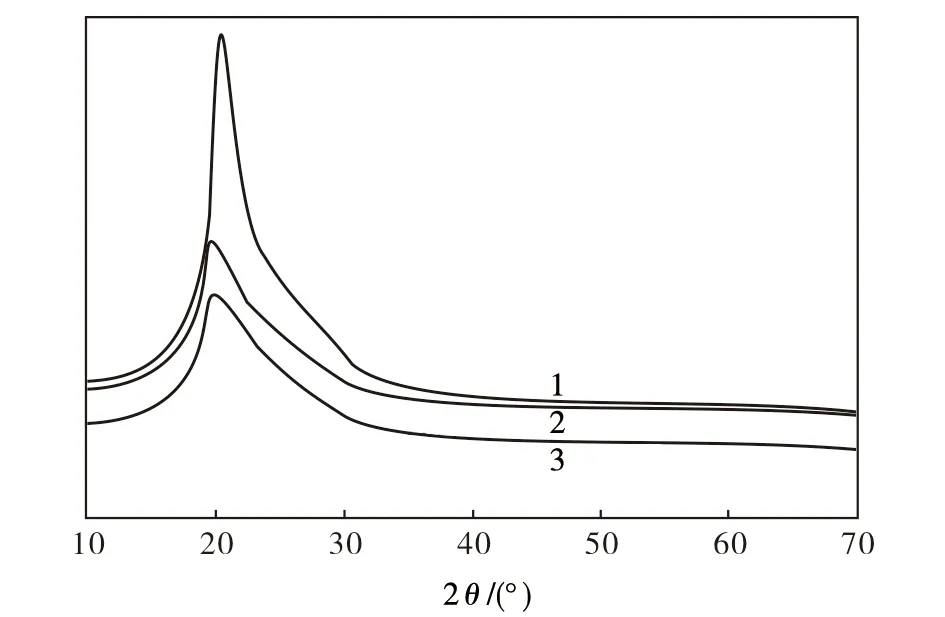
图4 试样的XRD图谱Fig.4 XRD spectra of samples1—F3;2—HCl-PANi/F3;3—PTSA-PANi/F3
2.4 PANi在F3表面的原位聚合过程及机理
根据Irina Sapurina等提出的PANi生长模型[25]及本研究涉及的PANi/F3产物制备过程,分析提出了如图5所示的PANi在F3表面的原位聚合过程机理。

图 5 PANi在F3表面的原位聚合过程示意Fig.5 In-situ polymerization process of PANi on F3 surface

2.5 吸波性能
F3和两种PANi/F3产物的复介电常数实部(ε′)、复介电常数虚部(ε″)、介电损耗角正切值(tanδε)与f的关系曲线见图6。

图 6 试样的ε′-f曲线与ε″-f曲线和tan δ ε-f曲线Fig.6 ε′-f, ε″-f and tan δ ε-f curves of samples●—HCl-PANi/F3;▲—PTSA-PANi/F3;■—F3
由图6可知:f在8~12 GHz,原料F3的ε′在2.70~2.82,而PTSA-PANi/F3、HCl-PANi/F3的ε′分别在3.0~3.1和3.00~3.15(见图6a);原料F3的ε″在0.05~0.28,而PTSA-PANi/F3、HCl-PANi/F3的ε″分别在0.20~0.37和0.40~0.58(见图6b),与原料F3相比较,在其表面原位合成PANi后,ε′和ε″都有一定程度增大,说明PANi在F3表面的原位合成使得F3的介电损耗性能得到了提升;原料F3的tanδε在0.01~0.10,而PTSA-PANi/F3、HCl-PANi/F3的tanδε分别在0.07~0.13和0.14~0.20(见图6c),与原料F3相比较,PTSA-PANi/F3和HCl-PANi/F3产物的tanδε均明显增大,表明两种产物较原料F3呈现出更为优异的介电损耗能力。
由图7可知,f在8~12 GHz,原料F3的复磁导率实部(μ′)在1.03~1.12,复磁导率虚部(μ″)近乎为0,磁损耗角正切值(tanδμ)最大值仅为0.05,表明其不具有磁损耗能力。而PTSA-PANi/F3、HCl-PANi/F3产物在与原料F3呈现相近的μ′值的同时,表现出明显高于F3的μ″和tanδμ值,表明两种PANi/F3产物较原料F3的磁损耗能力得到了明显提升。

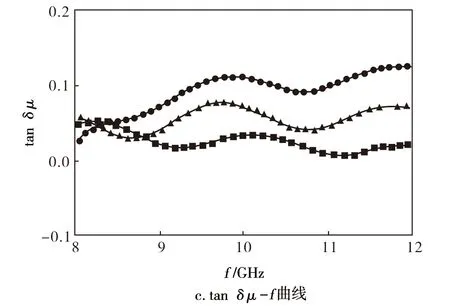
图 7 试样的μ′-f曲线与μ″-f曲线和tan δμ-f曲线Fig.7 μ′-f, μ″-f and tan δμ-f curves of samples●—HCl-PANi/F3;▲—PTSA-PANi/F3;■—F3
根据微波传输线理论可知,对单层吸波材料,入射到材料表面的电磁波输入阻抗(Zin)为[27]:
(1)
式中:μr=μr′-jμr″和εr=εr′-jεr″分别为复磁导率和复介电常数;自由空间阻抗(Z0)为337 Ω;d为材料厚度;c为光速。
RL的计算公式为[28]:
RL=20log|(Zin-Z0)(Zin+Z0)|
(2)
通过计算结果作出原料 F3和PTSA-PANi/F3、HCl-PANi/F3产物的RL随电磁波f和材料d而变化的三维色图,如图8示。
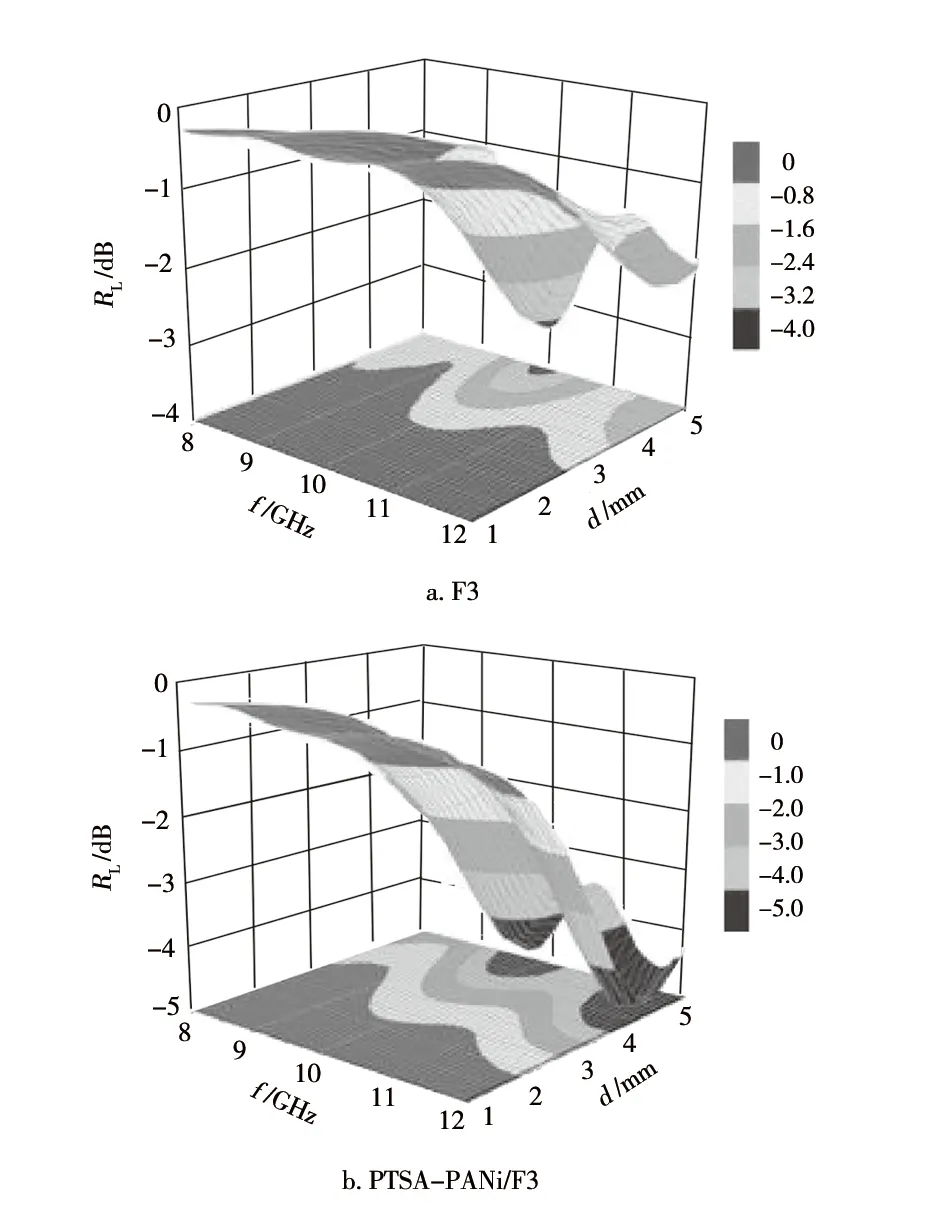
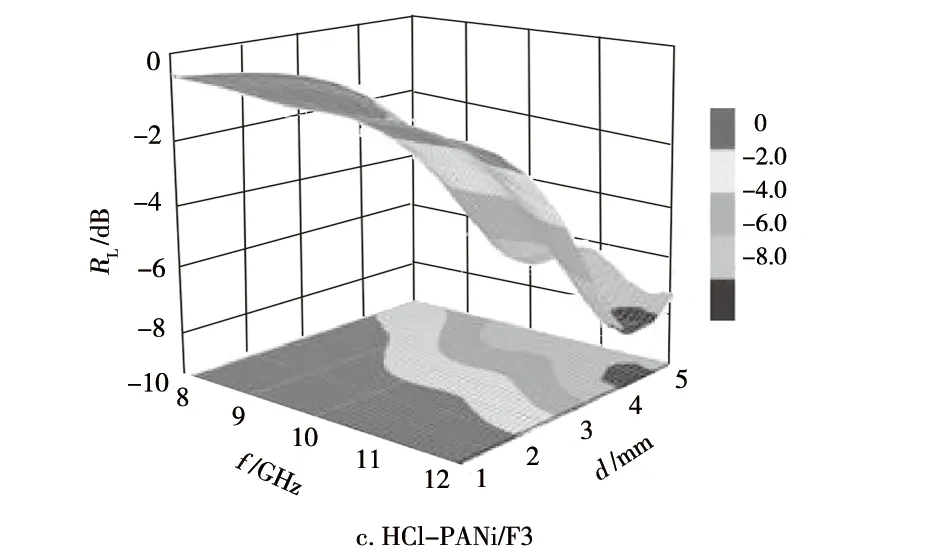
图8 试样的RL随f和d变化的三维色图示意Fig.8 Three dimensional color diagrams of RL versus f and d of samples
由图8可知:在f为8~12 GHz和d为1~5 mm时,F3无法获得优于-3.5 dB的吸波效果;PTSA-PANi/F3产物的最优吸波效果可达到-4.9 dB,优于-3.5 dB的频带范围,覆盖9.1~12 GHz;HCl-PANi/F3产物的最优吸波效果可达到-8.3 dB,优于-3.5 dB的频带范围,覆盖整个8~12 GHz。由此可知,相对于原料F3,在其表面原位合成PANi而制备的两种PANi/F3产物的吸波性能得到了显著的提升,其中,HCl-PANi/F3产物的吸波性能最为优异。结合本课题组前期研究结果,认为两种PANi/F3产物较原料F3所呈现的吸波性能显著提升,一方面归因于PANi在F3表面的原位负载导致PANi/F3产物的介电损耗、磁损耗能力较F3显著提升[13],另一方面得益于两种PANi/F3产物中F3与PANi膜层之间所形成的异质界面引起的较强的异质界面极化效应对入射电磁波的损耗作用[13]。就HCl-PANi/F3产物较PTSA-PANi/F3产物呈现更优的吸波性能,初步分析认为这与前者产物中的PANi氧化程度更高、导电性能更优(HCl-PANi/F3的电导率为2.36×10-3S/cm,PTSA-PANi/F3的电导率为2.12×10-4S/cm),以及其负载于F3表面的PANi膜层更为均匀致密有关。
3 结论
a.采用原位合成方法,实现了PANi膜层在F3表面的原位聚合与负载,制备了PTSA-PANi/F3和HCl-PANi/F3两种复合产物;PANi膜层与F3之间除了物理吸附作用以外,还存在氢键结合;PANi在F3表面的负载未对F3的结晶结构产生明显影响;由于氧化程度不同,两种PANi/F3产物呈现出一定的颜色差异,PTSA-PANi/F3呈墨绿色,HCl-PANi/F3呈灰黑色。
b.PANi在F3表面的生长机理为按有序刷子状方式沉积而形成膜层结构。
c.相对于原料F3,两种PANi/F3产物的介电损耗和磁损耗能力都得到了明显的提升。在8~12 GHz频段,两种产物均表现出优于F3的吸波性能,其中HCl-PANi/F3产物的吸波性能最为优异。
