支链氨基酸在非酒精性脂肪性肝病发生发展中的作用
2022-03-18谢小青刘亚贤于晓辉
谢小青, 刘亚贤, 陈 顺, 于晓辉
1 中国人民解放军联勤保障部队第九四〇医院 消化内科, 兰州 730050; 2 兰州大学第二医院 消化内科, 兰州 730030
非酒精性脂肪性肝病(nonalcoholic fatty liver disease,NAFLD)是慢性肝病最常见的病种之一,全球发病率为25%[1],我国为5%~24%,目前其发病率还在逐年上升[2]。NAFLD定义为>5%的肝细胞存在脂肪变性,且没有显著的持续或近期饮酒和其他已知的肝脏疾病原因[1]。NAFLD的组织学范围从单纯性脂肪变性到非酒精性脂肪性肝炎(nonalcoholic steatohepatitis,NASH)、肝纤维化和肝硬化[3]。NAFLD发生和发展的机制较复杂,有学者认为NAFLD发病机制可能与氨基酸代谢异常有关,其中支链氨基酸(branched chain amino acid,BCAA)已成为NAFLD的独立危险因素,而其代谢异常可引起机体产生一系列的代谢紊乱,这是导致NAFLD发生发展的关键因素。
BCAA包括亮氨酸、异亮氨酸和缬氨酸3种氨基酸,是人体所需的必需氨基酸,其分解代谢最初是在肝外组织中通过支链氨基酸氨基转移酶(branched chain amino acid transaminase,BCAT)和支链α-酮酸脱氢酶(branched chain ketoacid dehydrogenase,BCKDH)催化下,经过BCAT转氨和BCKDH脱羧后,通过一系列酶促反应转化为乙酰CoA和琥珀酰CoA,进入三羧酸循环(tricarboxylic acid cycle,TCA)完全氧化分解[4]。其代谢过程涉及胰岛素信号转导、脂肪酸氧化、TCA、糖酵解及线粒体氧化等多种生理过程。因此任何环节异常都可能导致NAFLD的发生,故本文对近年来的相关文献进行系统综述,以归纳总结BCAA在NAFLD发生与发展中的作用机制,为进一步深入研究提供理论依据。
虽然2020年国际专家共识将NAFLD定义为代谢性脂肪性肝病(metabolic fatty liver disease,MAFLD)[5-6],但共识指出MAFLD与饮酒量无关,且尚无MAFLD的具体分类,加之既往的文献中均以NAFLD进行研究报道,故本综述仍采用NAFLD这一关键词。
1 BCAA与肝细胞氧化应激
1.1 促炎因子 BCAA可刺激促炎基因的表达,导致促炎分子的释放,实验表明补充BCAA组小鼠与高脂饮食组相比,显著上调TNFα、IL-6、IL-1β和MCP-1的水平。
1.2 NF-κB信号 补充BCAA组上调NF-κB,增加人外周血单核细胞和内皮细胞,促进致炎因子释放、炎性细胞黏附和内皮功能障碍,加剧对肝细胞的损伤[7-8]。
1.3 脂肪合成和游离脂肪酸(free fatty acid,FFA) 补充BCAA会增加肝脏脂肪的从头合成,促进脂肪变性,还可使FFA蓄积引起氧化应激和脂质过氧化,进一步损害肝脏的结构和功能,导致NASH的发生。在高BCAA饮食中,脂肪生成的标志物明显增加,如ATP柠檬酸裂解酶(ACLY)、脂肪酸合成酶(FAS)和乙酰辅酶a羧化酶(ACC)。此外,高BCAA可能激活BCKDK,增加ACLY磷酸化,加强脂肪酸的从头合成[9](图1)。单磷酸腺苷活化蛋白激酶(adenosine monophosphate activated protein kinase,AMPK)是体内的“细胞能量感受器”,它决定身体中的脂肪是储存还是燃烧,激活后可明显促进脂解和脂肪酸氧化[10]。研究表明给小鼠补充BCAA可以显著促进AMPK的激活,并通过AMPKα2途径抑制磷酸二酯酶活性和促进激素敏感性脂肪酶活化,进一步增强脂解作用,导致FFA释放,加重肝脏损伤[11]。对112例NAFLD患者行超声、MRI和生化参数分析评估肝脏,表明BCAA摄入越多,肝脏状态越差,且BCAA与脂肪含量呈正相关[12]。然而在NAFLD的不同阶段,BCAA扮演的角色不一致,研究[13]认为其有明显的抗氧化作用,无论单独给予BCAA还是联合其他抗氧化物质均能减少脱氧核糖核酸损伤,抑制NASH的进展。
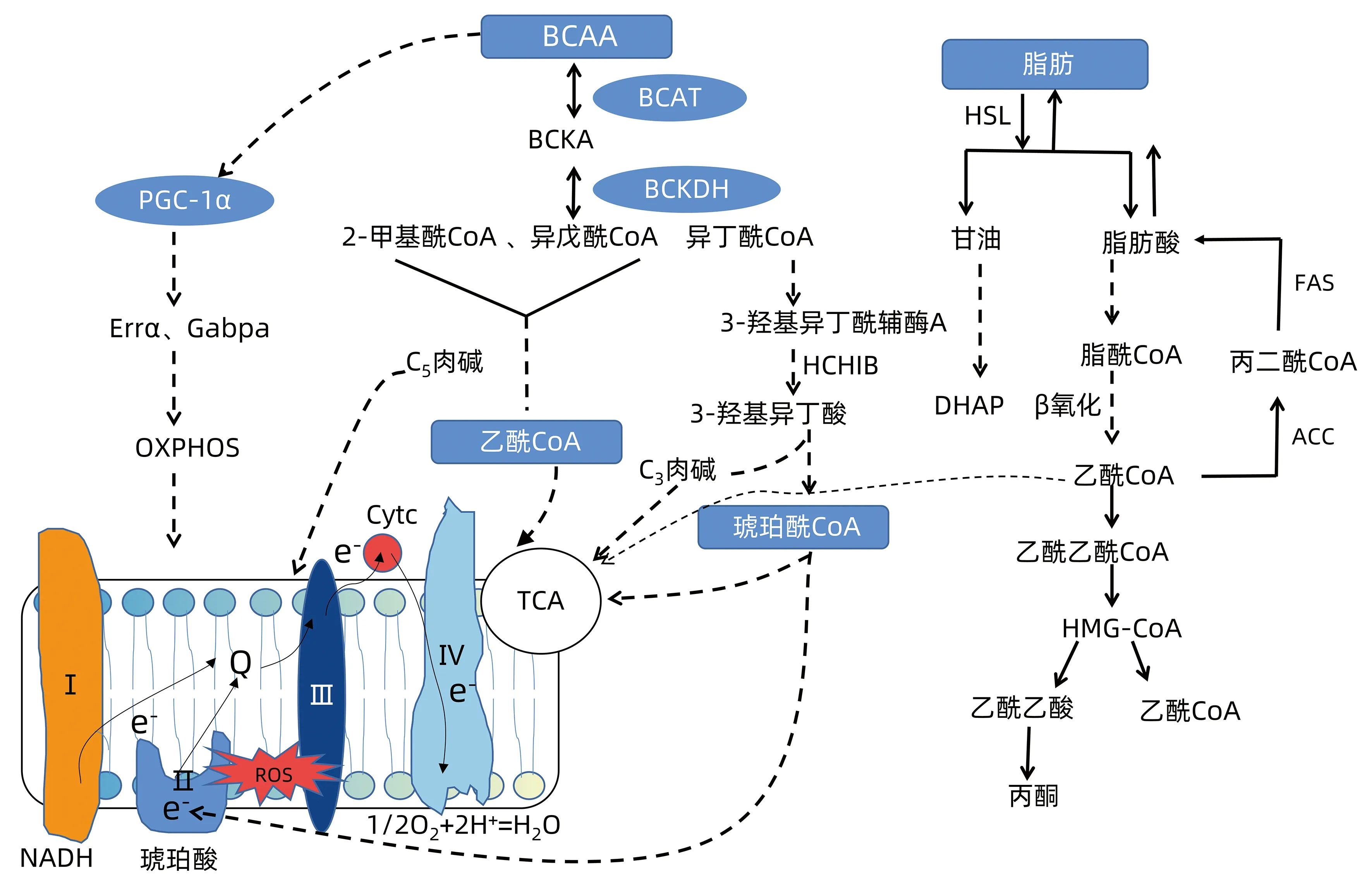
注:BCAA在BCAT的作用下生成相应的支链α-酮酸(BCKA),BCKA在BCKDH催化生成2-甲基酰CoA 、异戊酰CoA 、异丁酰CoA,经一系列酶促反应生成乙酰CoA,最终进入TCA,经过复合体Ⅰ(NADH)、Ⅱ、Ⅲ、Ⅳ氧化磷酸化后(OXPHOS)生成ATP和水。Cytc,细胞色素酶;Q,辅酶Q;HSL,激素敏感性脂肪酶;DHAP,3-磷酸甘油;HMG-CoA,羟甲基戊二酸单酰辅酶A;PGC-1α,过氧化物酶体增殖物激活受体-γ共激活因子1;Errα,雌激素相关受体α;Gabpα,GA结合蛋白α。
2 BCAA与肝细胞自噬
自噬作为细胞管家,清除损伤的肝细胞是机体的保护过程。BCAA诱导的小鼠中LC3-Ⅱ表达减少和肝自噬蛋白p62表达增加[11],具体的机制尚不明确,但是存在一些可能的假说:哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)是一种丝氨酸/苏氨酸激酶信号复合物,在自噬中扮演核心角色,BCAA激活mTOR信号显著抑制肝脏自噬,加重肝细胞的脂质暴露,损伤肝细胞[14-15]。乙酰CoA通过乙酰化调节mTORC1活性,也从侧面说明BCAA可影响自噬[16]。虽然目前BCAA关于自噬的研究匮乏,但已有大量文献证明BCAA激活mTORC1信号途径,抑制心肌和骨骼肌细胞中的自噬[17-18]。
3 BCAA与线粒体功能障碍
高BCAA可能通过PGC-1α增强了Errα和Gabpa的表达,进而激活了下游OXPHOS相关基因的表达,影响氧化磷酸化。此外,分解BCAA的大部分酶都在线粒体中,当代谢过程出现异常时,其代谢产物的堆积也可以反映线粒体的功能。
3.1 BCAA代谢产物 Peng等[19]对58例NAFLD和NASH患者肝脏和血浆脂质组学分析,发现肝心磷脂和泛醌在NAFLD患者中积累,酰基肉碱在NASH中显著累积。心磷脂可以促进复合物 Ⅰ 与泛醌之间的电子传输,心磷脂和泛醌不成比例的改变可能会形成不利于最佳电子传输的环境[20]。酰基肉碱是一种中间体,通过线粒体内膜运输后,释放附着的脂肪酰基进行β氧化,故其累计间接提示线粒体功能障碍和脂肪酸氧化受损[21-22]。
3.2 三羧酸循环(TCA) BCAA分解代谢后生成琥珀酰CoA进入TCA,琥珀酸脱氢酶在TCA中催化琥珀酸到富马酸的转化,也是电子转移链中的呼吸复合物Ⅱ。在肝细胞中,BCAA代谢异常所致BCKA升高可以抑制琥珀酸脱氢酶的基因表达,从而阻断TCA和ATP的产生[23]。
3.3 生酮环境 代谢组学显示NAFLD患者肝脏BCAA水平升高[24],而长期高BCAA会导致肝细胞长期暴露于生酮的环境中,显著诱导生酮基因Hmgcs2和Bdh1表达。肝细胞将更多的乙酰CoA从脂肪合成转到生酮和TCA,不仅引发促炎信号级联,还促进慢性ROS的产生和肝细胞应激,加速氧化应激和炎症环境,进一步加重肝线粒体功能障碍[25]。
4 BCAA与胰岛素抵抗
胰岛素是调控机体葡萄糖和脂质代谢的关键激素。生理条件下,机体分泌胰岛素与相应受体结合,促进胰岛素受体(IRS)的酪氨酸磷酸化并调节下游级联信号。NAFLD患者中BCAA升高与胰岛素抵抗密切相关[26],然而影响胰岛素敏感性和糖代谢的具体机制有待进一步探讨。目前主要存在两种模型,一种是mTORC1信号通路模型: BCAA激活mTORC引起胰岛素抵抗。可能的机制有:(1)大鼠骨骼肌中亮氨酸水平升高抑制AMPK活化,并激活了mTOR / S6K1信号导致胰岛素抵抗[27],此外,降低亮氨酸可激活GCN2(一种氨基酸传感器)和降低mTOR信号来改善肝胰岛素敏感性。(2)BCAA会促使机体产生FFA,FFA可调节IRS蛋白磷酸化或通过组蛋白乙酰化的IRS转录水平影响IRS的活性[28]或者通过FFA→PKCδ→NADPH氧化酶和氧化应激→IKKβ/ JNK→肝胰岛素信号传导受损途径[29](图2)。(3)实验证明暴露于高水平BCKA可激活mTORC1抑制胰岛素诱导的AKT磷酸化,并用mTORC1抑制剂干预肌肉细胞后可恢复该作用反向验证信号通路的正确性,强调了BCAA在调节肌肉胰岛素信号传导中的生物学作用[30]。第二种机制是代谢紊乱模型:它认为BCAA不是胰岛素抵抗的直接原因,但胰岛素抵抗是由BCAA分解代谢紊乱产生的有毒代谢中间体引起的,这些代谢中间体可以损害细胞功能并诱导胰岛素抵抗,这种模式目前被高度接受。在骨骼肌中,缬氨酸产生的3-HIB促进骨骼肌脂肪酸摄取,导致骨骼肌中不完全氧化的脂质和血浆中的BCKA积聚,引起胰岛素抵抗[30]。在肝脏中,BCKA大量分解,导致多种酰基肉碱积累,破坏线粒体TCA,造成不完全氧化产物积累和胰岛素抵抗。在脂肪组织中,BCAT和BCKDH的表达减少,导致血浆BCAA水平升高,从而引起骨骼肌和肝脏等组织中BCAA的大量分解,并诱导胰岛素抵抗。
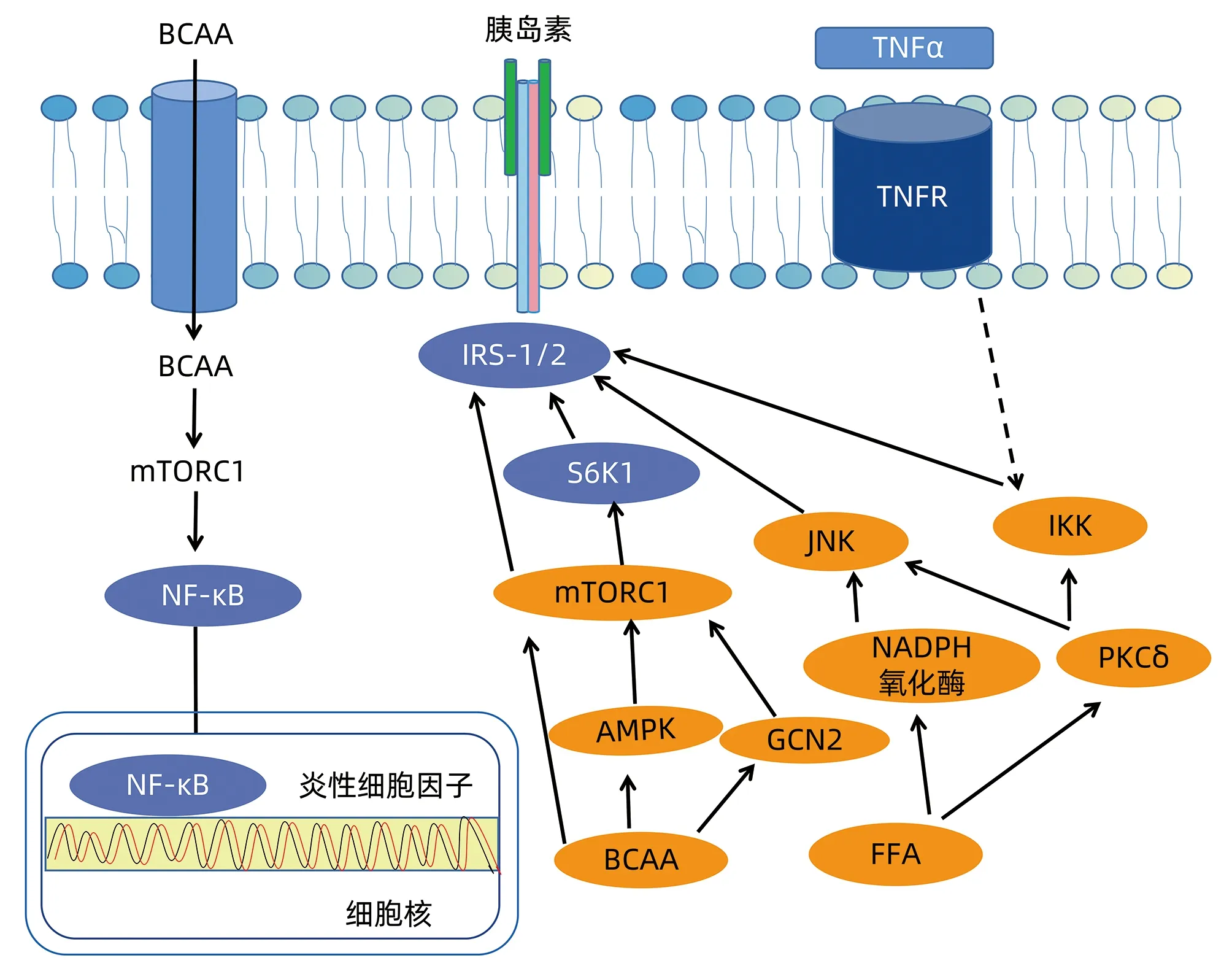
注:S6K1,核糖体蛋白S6激酶1;GCN2,阻遏蛋白激酶2;JNK,c-Jun氨基末端激酶;IKK:kappaB激酶。图2 BCAA与氧化应激及胰岛素抵抗信号通路
5 BCAA与摄食行为
色氨酸是产生5-羟色胺的唯一前体,BCAA可通过血流进入大脑,降低5-羟色胺生成,其作为机体的一种血清素是控制食欲的单胺类神经递质,含量越低,越促进食欲。故推测长期高BCAA摄入对机体的影响不一定是由内在毒性引起的,而是氨基酸失衡驱动摄食过度的结果,特别是色氨酸的变化,且与中枢血清素耗竭有关。高BCAA小鼠血浆中色氨酸、5-羟色胺及其主要代谢物5-羟基吲哚乙酸均减少。以BCAA200或BCAA100饮食喂养小鼠6周,使用局部电刺激诱发5-羟色胺的突触释放。BCAA100小鼠中诱发抑制性突触后电流(IPSC),该电流可被5-羟色胺受体1A受体拮抗剂阻断。BCAA200小鼠在刺激时表现出明显钝化。与BCAA100小鼠相比,BCAA200小鼠的平均诱发IPSC振幅显著降低,向培养基中添加色氨酸可阻断其反应[31]。Dyrk1a、Ttc7b和Nlrp3三个下丘脑基因分别与食欲、肥胖和炎症相关。动物实验[32]表明高BCAA摄入量与这些基因呈强正相关。下丘脑Dyrk1a信使核糖核酸表达增加的转基因小鼠表现出食物摄入增加,由叉头盒氧诱导的Npy(食欲调节途径中的一个关键基因)表达介导。Ttc7b表达是脂肪细胞功能和脂质储存的标志,随着BCAA摄入量的增加以及炎症体标志Nlrp3的增加而升高。
6 小结
总之,BCAA水平升高通过影响线粒体功能和炎症反应进而引起NAFLD,而线粒体功能障碍,氧化应激和胰岛素抵抗可能会进一步导致BCAA蓄积,从而导致NAFLD的发生发展。但是,BCAA是否通过其他信号途径协同引起NAFLD?BCAA异常水平是否可以作为风险预测的有用标志物?BCAA能否做为NAFLD疾病中的新靶点在临床应用和推广?这些问题都需要进一步研究。
利益冲突声明:所有作者均声明不存在利益冲突。
作者贡献声明:谢小青负责课题设计,撰写论文;刘亚贤、陈顺参与修改论文;于晓辉负责拟定写作思路,指导撰写文章并最后定稿。
