超高效液相色谱-串联质谱法测定藻类保健食品中节球藻毒素
2022-03-17李硕,李莉
李 硕,李 莉
(1.中国计量科学研究院,北京 100029;2.中国食品药品检定研究院,北京 100050)
节球藻毒素(Nodularin,NOD)是由泡沫节球藻(Nodularia spumigena)产生的具有强烈肝毒性的蓝藻毒素,具有强烈的基因毒性、胚胎毒性和遗传毒性,长期接触会增加癌症患病的风险[1-6]。节球藻毒素为环状五肽,分子结构式见图1。国际癌症研究机构(International Agency for Research on Cancer,IARC)公布的致癌物分级清单将节球藻毒素列为3类致癌物[7]。

图1 节球藻毒素的结构式Fig.1 Molecular structure of nodularin
近年来,以螺旋藻为代表的藻类保健食品,因富含蛋白质、氨基酸、多糖、膳食纤维、不饱和脂肪酸、矿物元素及多种生理活性物质,日益受到消费者的关注和青睐[8-9]。除螺旋藻外,我国目前已批准的藻类保健食品原料还包括小球藻、盐藻、雨生红球藻等[8]。由于可用于保健食品原料的藻类养殖水域可能有其他藻类(如微囊藻、节球藻等)与其共生,因此这些保健食品原料存在被其他藻类所产生毒素污染的风险[8,10]。此外,节球藻毒素化学性质稳定,即使加热煮沸都难以破坏其结构,在藻类保健食品加工过程中,如果质量控制不严格,很有可能将节球藻毒素传递至终产品,对消费者身体健康造成潜在危害[10-12],存在潜在安全风险。截至目前,我国尚未出台食品中节球藻毒素的限量标准,GB 19643-2016《食品安全国家标准 藻类及其制品》[13]、GB/T 16919-1997《食用螺旋藻粉》[14]均未规定节球藻毒素的检验项目。为有效开展藻类保健食品中的安全风险评估,准确识别保健食品中节球藻毒素的污染程度,有必要建立藻类保健食品中节球藻毒素含量的测定方法。
目前节球藻毒素的测定分析方法主要包括酶联免疫吸附测定法[15-16]、高效液相色谱法[17-18]、液相色谱-质谱法[18-31]等方法,主要集中在水质监测等环境领域[18-27]以及水产品安全领域[28-32],有关藻类保健食品中节球藻毒素污染情况的研究较少[26-27],检测方法灵敏度较差。本研究采用固相萃取样品前处理技术,并结合高效液相色谱-串联质谱技术,建立藻类保健食品中节球藻毒素的检测方法。该方法旨在建立简便、快速、灵敏度高的检测方法,对了解藻类保健食品中节球藻毒素污染水平及潜在安全风险具有重要意义。
1 材料与方法
1.1 材料与仪器
节球藻毒素标准溶液(10.21 μg/mL) 青岛普瑞邦公司;甲醇(色谱纯)、乙腈(色谱纯) 美国Fisher Scientific公司;甲酸(质谱纯) 美国Sigma公司;Captiva EMR-Lipid Cartridge(3 mL) 美国 Agilent公司;DisQuE tube(含 150 mg 硫酸镁、25 mg PSA、30 mg C18和30 mg中性氧化铝) 美国Waters公司;SHIMSEN QuEChERSdSPE column(含 150 mg硫酸镁、25mg PSA、25 mg C18) 日本Shimadzu公司;螺旋藻及小球藻等藻类保健食品,包括片剂、胶囊和藻粉 均购自网络平台。
LCMS-8060型超高效液相色谱-三重四级杆串联质谱仪配有电喷雾离子源(ESI)及LabSolutions(Version 5.99 SP2)数据处理系统 日本Shimadzu公司;AL 204型分析天平和XP205型分析天平 瑞士Mettler Toledo公司;Vortex Genie 2涡旋混匀器美国Scientific Industries公司;GTR21-1B医用离心机 北京新时代北利医疗器械有限公司;Arium®pro VF超纯水纯化系统 德国Sartorius公司。
1.2 实验方法
1.2.1 提取 样品称量前根据不同剂型进行初步处理,其中固体片剂用杵和研钵磨成细粉末;胶囊打开后倒出内部粉末;藻粉无需做进一步处理。准确称取进行初步处理后的样品0.5 g(精确至0.0001 g)样品置于15 mL离心管中,加入5 mL提取溶液(甲醇:水=80:20,v/v),旋涡振荡 30 s,在 4 ℃ 条件下,8000 r/min离心5 min。
1.2.2 净化
1.2.2.1 净化柱净化 取1.2.1提取步骤中所得上清液 1 mL,经 Captiva EMR-lipid 净化柱净化,0.22 μm聚四氟乙烯微孔滤膜过滤,续滤液作为待测溶液。
1.2.2.2 净化管净化 取1.2.1提取步骤中所得上清液1 mL,置于DisQuE或SHIMSEN QuEChERSdSPE净化管中涡旋混合1 min,在4 ℃条件下,8000 r/min离心5 min。上清液经0.22 μm聚四氟乙烯微孔滤膜过滤后待测。
1.2.3 标准溶液的制备 取节球藻毒素标准溶液适量,用50%的甲醇水溶液配制成质量浓度为1 μg/mL的标准储备溶液。将标准储备液稀释成质量浓度为100 μg/L的中间标准溶液。取中间标准溶液适量置于10 mL容量瓶中,用50%的甲醇水溶液稀释得到质量浓度分别为 0.5、1、5、10、25和 50 μg/L的标准系列工作溶液。
1.2.4 仪器条件
1.2.4.1 色谱条件 色谱柱:Agilent Infinity Poroshell 120 SB-C18(100 mm×2.1 mm,2.7 μm);流动相:A 为含有0.01%甲酸的水溶液,B为乙腈;梯度洗脱程序 :0~3 min,25%~50%B;3~5 min,50%~90%B;5~5.6 min,90%B;5.7~7 min,25%B;柱温:40 ℃;进样量:5 μL;流速:0.3 mL/min。
1.2.4.2 质谱条件 离子源:电喷雾离子源(electrospray ionization,ESI);正离子扫描模式;检测模式采用多反应监测(multiple reaction monitoring,MRM)模式;雾化气流量为 3 L/min,干燥气(氮气)流量为10 L/min,加热气(空气)流量为10 L/min,碰撞气为氩气,离子源接口温度为300 ℃,接口电压为4.0 kV。质谱采集时间为2.5~4 min。质谱参数见表1。
1.2.5 样品测定 通过采用本研究方法,对39批次螺旋藻及小球藻等藻类保健食品样品进行检测,所有样品均未检出节球藻毒素。
1.3 数据处理
实验数据通过岛津LabSolutions软件采集,导出原始数据后采用OriginPro 2016计算和绘图。
2 结果与分析
2.1 质谱条件的优化
配制质量浓度为50 μg /L节球藻毒素标准溶液,经质谱分析,得到目标化合物的母离子。通过对比目标化合物在正、负离子模式下的响应值发现,节球藻毒素在正离子模式下响应较高,因此选择正离子扫描模式。随后通过优化电压及碰撞能,选择响应最强的2对离子,其中m/z:825.4>135.2作为定量离子对,m/z:825.4>227.3作为定性离子对。优化后的节球藻毒素母离子、子离子及质谱参数见表1。

表1 节球藻毒素的质谱参数Table 1 MS parameters of nodularin
2.2 色谱条件的选择
由于选择正离子模式扫描,在流动相中加入适量的酸有助于提高目标物质的离子化效率,因此选择0.1%甲酸水溶液和乙腈作为流动相体系,梯度洗脱。样品经提取和净化处理后上机测定,不同净化方式下的节球藻毒素MRM谱图见图2。由图2可知,采用不同净化方式净化后测定,节球藻毒素色谱峰周围无明显干扰,说明该色谱条件选择性良好。

图2 不同净化方式下节球藻毒素MRM谱图Fig.2 MRM spectra of nodularin under different purification conditions
2.3 样品前处理条件的优化
2.3.1 提取溶剂和净化方式的优化 本研究比较了6种提取溶剂的提取效果以及3种净化方式的净化效果。加标样品分别采用甲醇溶液比例分别为50%(v/v)、60%(v/v)、70%(v/v)、80%(v/v)、90%(v/v)以及纯甲醇等6种提取溶剂提取。然后再分别经Captiva EMR-Lipid Cartridge、DisQuE tube和 SHIMSEN QuEChERSdSPE column 3种净化装置净化后上机测定,加标回收率结果如图3所示。
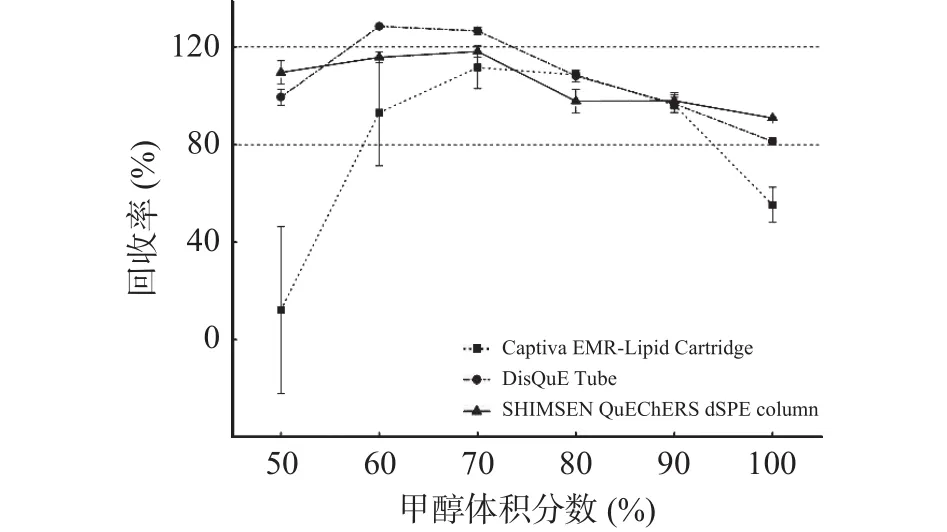
图3 提取溶剂对节球藻毒素回收率的影响(n=3)Fig.3 Effects of extraction solvent on recoveries of nodularin(n=3)
实验结果表明,不同净化装置对同一提取溶剂的净化效果存在较大差异:当采用SHIMSEN QuEChERSdSPE column进行净化时,所比较的6种提取溶剂均能取得较好的效果,满足回收率在80%~120%之间;当采用Captiva EMR-Lipid Cartridge进行净化时,随着提取溶剂中甲醇的比例上升,节球藻毒素的回收率先提高后降低;当采用DisQuE tube进行净化时,随着提取溶剂中甲醇的比例上升,节球藻毒素的回收率先提高后降低。当提取溶剂中甲醇的比例为80%(v/v)或90%(v/v)时,3种净化装置都能取得较好的效果,均满足回收率在80%~120%之间的需求。当仅考虑提取效果、净化效果以及方法净化装置的通用性时,提取溶剂中甲醇的比例为80%(v/v)或90%(v/v)均可作为候选的提取溶剂。但当甲醇的比例由80%(v/v)提升为90%(v/v)时,随着甲醇所占比例的增加,净化后的待测溶液颜色明显加深,进样后液相小瓶瓶盖留有绿色物质残留,表明随着提取溶液中甲醇比例增加,样品中更多的色素成分被提取出来进入待测液中。考虑到色素成分的增加不仅会增大对目标物质检测的干扰,而且还会污染进样装置和质谱系统,因此本实验最终选择80%甲醇水溶液(v/v)作为提取溶剂。在实际应用中,可以根据实验室所在单位对选择供应商的政策要求、不同净化方式操作熟练程度,以及不同厂家净化管(柱)的送货期、价格等因素灵活选择。
2.3.2 提取方式的优化 在空白基质中加入节球藻毒素标准储备溶液25 μL和提取溶剂后,分别采用涡旋提取30 s或超声5、10、20、30 min提取的方式进行提取,经Captiva EMR-Lipid净化管净化后上机测定,通过计算加标回收率考察不同提取方式的提取效率(结果见表2),并对各组数据进行方差分析。从结果可以看出,采用涡旋和超声不同时间提取的回收率均满足检测要求;方差分析计算得到F=1.346,Fcrit=3.478,F<Fcrit,表明几种提取方式之间无显著差异。但是实验中发现,随着提取时间的延长,样品提取溶液颜色变深,说明有更多的色素等杂质被提取出来。为了减少杂质对质谱的污染,试验最终选择采用涡旋30 s的提取方式。

表2 不同提取方法时节球藻毒素的回收率和精密度Table 2 Recoveries and precisions of nodularin in different extraction method
2.4 基质效应
采用质谱分析时,基质中的共提取物可能会对待测节球藻毒素仪器响应值产生抑制或增强作用,从而导致含量测定偏高或偏低,影响结果的准确性和可靠性。通过对比基质匹配标准曲线斜率(kM)与溶剂标准曲线斜率(kS)的相对值,可以考察样品基质对节球藻毒素定量分析结果的影响,期量化评价待测样品基质对待测组分的影响。当(1-kM/kS)×100%为正值时,则表示基质对待测组分存在抑制作用;当(1-kM/kS)×100%为负值时,则表示基质对待测组分存在增强作用;当(1-kM/kS)×100%为零时,则表示不存在基质效应。具体操作:称取空白藻粉样品,用提取液提取并经净化后用于配制节球藻毒素浓度为0.5、1、5、10、20和 50 μg/L 的基质匹配标准工作溶液;同时用甲醇配制相同浓度的标准工作溶液。分别进样测定、读取峰面积、拟合工作曲线并计算曲线斜率,可得基质匹配标准曲线的斜率(kM)数值与溶剂标准曲线的斜率(kS)数值分别为17188和17394。按照式(1)对基质效应影响进行评价:

计算结果表明,与纯溶剂配制的标准溶液相比,在空白样品基质溶液对节球藻毒素质谱响应值的抑制效应约为1.2%。基质干扰对节球藻毒素质谱检测影响较小,采用纯溶剂配制的标准工作曲线可以满足定量测定要求。
2.5 线性关系考察
分别取1.2.5中的系列标准工作溶液各5 μL按上述条件测定,以目标物质的峰面积为纵坐标(Y),相应浓度为横坐标(X),绘制得到标准曲线Y=17394X+13267。结果显示,节球藻毒素含量在0.5~50 μg/L范围内相关系数(r)为0.9997,线性关系良好。当样品称样量0.5 g时,以3倍信噪比(S/N=3)确定化合物的检出限(limit of detection,LOD)为 1 μg/kg、10倍信噪比(S/N=10)确定化合物的定量限(limit of quantification,LOQ)为 3 μg/kg。
2.6 加标回收率与精密度
分别在不含节球藻毒素的螺旋藻粉中,添加10、50和250 μg/kg低、中、高3个浓度水平的节球藻毒素标准溶液进行测定,计算平均回收率(n=6)及相对标准偏差(relative standard deviations,RSD),结果见表3。结果表明,节球藻毒素在3个添加水平下的平均回收率范围在103.6%~114.8%之间,相对标准偏差为3.3%~12.5%,从整体上看,该方法准确度和重现性良好,能够满足日常检测要求。

表3 不同添加水平下节球藻毒素的回收率和精密度(n=6)Table 3 Recoveries and precisions of nodularin at three different levels (n=6)
3 结论
本研究采用固相萃取方法对样品进行净化,并结合UPLC-MS/MS技术,建立了藻类保健食品中节球藻毒素的检测方法,通过对提取溶剂和净化条件的优化,样品经甲醇-水(80:20, v/v)混合振荡提取,经净化柱或净化管净化,在优化后的仪器条件下,节球藻毒素在线性范围0.5~50 μg/L上具有良好的线性关系,检出限为 1 μg/kg,定量限为 3 μg/kg,在低、中、高3个浓度下的平均加标回收率为103.6%~114.8%,相对标准偏差为3.3%~12.5%。通过应用本方法对39批市售藻类保健食品样品进行检测,所有样品均未检出节球藻毒素。该检测结果表明:我国藻类保健食品中节球藻毒素的污染风险水平较低,整体较为安全。本方法操作简便、灵敏度高、定量准确、重现性好,可以实现批量样品的检测,适合藻类保健食品中节球藻毒素的定性确证与定量检测。
