基于第一性原理对于锂硫电池正极材料NiS掺杂改性的研究
2022-03-13王自立
吴 宇,王自立,邓 超
(哈尔滨师范大学)
0 引言
传统锂硫电池一般采用硫单质作为正极,金属单质作为负极,而传统的正极材料单质硫是不导电物质,导电性能非常差且在充放电过程中,体积的扩大或缩小的程度非常大可能会导致电池的损坏.传统锂硫电池导电性差,多硫化物溶解严重,制约其应用与发展,而过渡金属具有优越的导电性,其硫化物更是有良好的界面相容性、理论比容量高、电压适中、电子导电性相对较高,被认为是高性能全固态锂电池极有前途的正极材料[1].Long等学者验证其合理性,但是,对于这种电极材料的电化学特性,特别是在分子层面,很多学者还没有足够的认识[2].因此,必须全面理解多硫化物在电池中的界面相互作用,才能更好地解释其在电池中的吸附.N(或 P等)掺杂提高了电极材料的电化学性质,提高了其导电率,提高了 Li+的迁移能力.从理论上论证了这种方法的合理性.研究了 N/NiS对Li2S4的S—S键的影响,并对其催化转化进行了研究.研究发现,氮化镍硫能够有效地控制电子态向费米能级运动,并改善其导电性能,从而对电极的电子动态进行调控.
1 计算方法
基于密度泛函理论和平面波赝势法进行了理论计算采用Perdew-Burke-Ernzerhof (PBE)交换相关函数的广义梯度近似(GGA),平面波截止能量设为400 eV.所有的几何优化和能量计算都使用周期边界条件进行.相邻分子与平板之间的距离至少为15 Å[3].而且倒数空间只使用了Γ点.收敛判据设定为残余力小于0.01 eV/Å,总能量变化小于10-6eV.结合能可以表示为E(b) =E(Na2Sx) +E(slab)-E(Na2Sx@slab),其中E(Na2Sx@slab)、E(Na2Sx)和E(slab)分别为吸附体系、Na2Sx种和表面板的总能[4].
2 结果与讨论
2.1 吸附构型与吸附能
在现有的资料中,石墨烯对锂硫化合物的吸附能力较差,其吸附能仅为- 0. 341~ - 0. 584 eV,吸附能较低表明,石墨烯材料并不能很好的吸附锂硫化合物[5].
利用DFT计算,通过NiS与N/NiS对锂硫化合物的吸附能的分析可知,N掺杂NiS对多硫化物有着较高的吸附能,此外,Li2S4、Li2S2等多硫化物的吸附能力明显高于Li2S6、Li2S.为深入探讨 NiS与 N/NiS的差别,该文对多硫化物在 NiS上的吸附行为进行了比较.该文对Li2Sx(x=1,2,4,6)在 N/NiS上的吸附形态进行了研究,图1显示了稳定的吸附构型,并且对吸收能量进行了计算,如图 2所示.

图1 N/NiS吸附Li2S/Li2S2
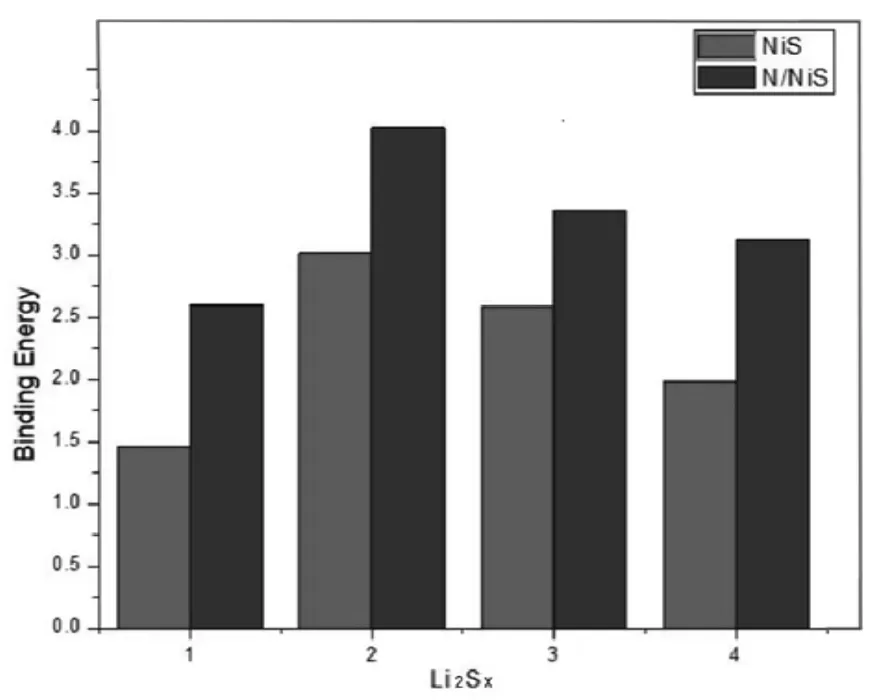
图2 N/NiS吸附Li2S/Li2S2吸附能
Li2Sx(x=1,2,4,6)吸附在NiS上,吸附能在 - 1. 455~ - 3.015 eV之间.而在N/NiS的吸附能在- 2. 598 eV ~ - 4.028 eV之间,N/NiS对锂硫化合物的吸附作用进一步的加强了,通过吸附数据,可以知道,N/NiS可能是一种理想的电极材料.
在进行结构优化后,可以从吸附锂硫化合物中的键长数据来进一步分析N/NiS作为正极材料的优越性,键长越短键能越高,经过结构优化可以很明显的发现,N/NiS与硫有着更短的键长,所以推测N对于NiS的掺杂增强了与多硫化物之间键的作用,从而增强了N/NiS自身的吸附作用.根据计算结果发现,N/NiS在对Li2S6和 Li2S4等多硫化物吸附后,因为N/NiS作为电极材料N/NiS与多硫化合物之间所形成的键长变短了.除此之外,Ni —S在N/NiS中可溶性多硫化物(Li2S6和 Li2S4)中的键长短于NiS吸附多硫化合物所形成的键长,更短的键长意味着更高的吸附能对锂硫化合物有着更高的催化作用,该反应能有效地抑制多硫化物的穿梭溶解,并使Li2S的降解速度降低,从而显著提高了催化剂的总体性能.
从根本上解释了过渡金属硫化物具有较高的吸附能.着重研究了Li2S4在放电时的键长以及在充电期间Li2S的变化.NiS对Li2S4的S—S键具有1.987 Å的长度,而在Li2S4中,S—S键的长度为1.798 Å.在Li2S键中,Li—S键由2.157 Å增加到 NiS的2.341 Å.由上述结果可知, N掺杂 NiS能较好地减弱 S—S和Li—S键的强度.N/NiS的吸附能比 NiS的高,这是因为 N的缺电性使得 N掺杂 NiS的极化能力较强.
根据上述的吸附能和键长数据,得到了如下结论:多硫锂的表面可能存在 N/NiS和 NiS.在放电期间, NiS与Li2S6分子在 N/NiS的同一时间上发生了强烈的 NiS键,并在后续的反应中逐步转变成Li2S4.Li2S4与 N/NiS形成了很强的Ni—S键,但随着S—S桥键的延长,Li2S4的键能逐渐降低.随着S—S键的进一步断裂和多硫化锂的转化,最后生成Li2S.在电荷作用下,硫化锂与氮化镍之间形成了强的Ni—S键,使Li—S键易于伸展、变弱,最终分解为硫化物.
2.2 总态密度
为了多方面探究过渡金属硫化物作为正极材料的优越性,对态密度进行了分析.研究不同种材料对态密度的影响,对费米能级处的情况进行分析.如图3所示,经过对比可以知道,N/NiS对于NiS来说具有更低的带隙的宽度,两者在费米能级处都处于电子态密度中,两者均具有金属性质[6].从理论考虑认为经过N掺杂的NiS有着更好的金属性和缺电子性,促使费米能级向着更高能级移动,证明体系自身具有良好的导电性,可以作为一种优秀的电极材料.
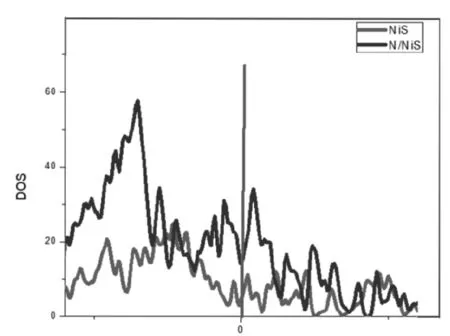
图3 N/NiS和NiS总态密度
N的掺杂使NiS产生了新的化学键,新的电子密度的产生使带隙降低,电子跃迁所需要的能量降低,表现出良好的导电性,提高电导率.
2.3 离子扩散能垒
锂离子的脱嵌会对电池自身的倍率产生直接的影响.在正极材料中,锂离子的脱嵌会因电荷的作用而产生极化,从而对离子的迁移产生一定的影响.由此,对锂离子的迁移进行了数值模拟,并对扩散期间的能量进行了分析.对 Li+在 NiS和 N/NiS上的扩散能垒进行了计算.Li+在 N/NiS的扩散能垒只有0.37 eV,与 NiS(0.68 eV),通过对比发现, N/NiS的扩散能垒较小,Ni—N键的生成对 Li+的扩散更为有利.
将氮化物掺杂到 NiS中,可以制备出一种具有 N/NiS双重功能的新型催化剂.N/NiS可以加速多硫化物在放电时的转化和在充电时的氧化.从理论上看, N的掺杂可以使硫化物的电子结构发生变化.DFT分析也表明了双官能界面上硫的转化机制.这说明,通过对含氮的双功能催化剂的研究,可以有效地改善锂离子电池的电化学特性.通过研究离子掺杂对催化剂的电子结构及配合环境的调节,可以为开发新的催化剂,开发出更好的锂电池.
2.4 晶体场理论
按照晶体场理论,一个正八面体的晶体场,会使过渡金属的d轨道分裂为低能级的三个兼并的低能级dxy,dyz,dxz,这三个兼并的低能级轨道统称为非键态的t2g态,和两个兼并的较高能级 ,这两个较高的兼并能级统称为反键态的eg态.NiS可以看作是低自旋半导体,从他们的总态密度图可以得知.至于NiS键合方式的描述,早在1934年就已经存在由Pauling和Huggins提出的共价的关系描述.包括Ni上的dsp轨道杂化与S上的sp3轨道杂化的交叠,这种杂化后来得到了各种电子结构计算的支持.这符合局域化的3d电子优先占据t2轨道的观点.
3 结论
利用密度泛函理论对锂离子电池正极材料N/NiS和NiS对多硫化合物的吸附,脱嵌,以及结构进行研究.从能量角度来说N掺杂的NiS,N与金属Ni形成新的Ni—S键,使N的周围形成新的催化中心,使得在吸附过程中有着更高的吸附能,以及更低的扩散能垒,使得锂离子更容易发生吸附和脱嵌且提高电池本身的倍率性能,表现出更强的金属性导电性.从键能的角度来考虑,N的掺杂使Ni—S键变短,提高了吸收多硫化物的能力,具有更高的能量.在多硫化物中, S—S键的延长,有利于多硫化物的转化.
