石油分子重构技术的研究进展
2022-03-11胡元冲褚小立李明丰
田 旺, 秦 康, 胡元冲, 褚小立, 李明丰
(中国石化 石油化工科学研究院,北京 100083)
石油是一种储量有限且十分宝贵的能源,对国民经济的发展具有重要的战略意义。充分挖掘石油的价值,将其高效利用,一直是石化行业研发人员关注的热点问题。对石油产品而言,其分子组成决定其物理和化学性质,也决定了其应用市场、价值。因此,为了合理利用石油资源,提高其在石油炼制领域的加工和转化效率,获得高价值的石油产品,有必要认识石油的分子组成信息,进而有针对性地为炼油过程开发出相应的催化剂和炼化工艺。
石油是由大量烃类和非烃类组成的混合物,不仅分子种类众多,而且存在较多的同分异构体,想要弄清其分子结构,十分困难。尽管如此,无数分析工作者做出了大量的努力,开发出多种色谱和高分辨质谱技术,对石油分子结构的认识逐渐深入。结合一系列分离技术,通过全二维气相色谱质谱(GC×GC/MS)、气相色谱-质谱联用仪(GC/MS/MS)、气相色谱飞行时间质谱(GC/TOF MS)以及傅里叶变化离子回旋共振质谱(FT-ICR MS)等分析手段,能得到烃类、非烃类化合物等石油馏分按碳数分布或者沸点分布的信息[1]。但是,即使是目前最先进的分析技术,依然无法获取石油中汽油以上馏分的详细分子组成。为了在现有分析数据的基础上,仍能得到必要的分子组成信息,经过大量研究发现[2-4],石油中分子结构基团以同系物的形式存在,并且呈现一定的分布规律。这一特点,一方面使轻馏分中结构基团的分布规律可以延伸到重馏分中;另一方面使重馏分中庞大数量的分子可以进行合理的归类和集总,使归类后的分子集在模型构建中的应用成为可能。研究人员将石油结构基团的分布规律和先进的数学工具结合,开发出了石油分子重构的技术路线。
石油分子重构技术是一项化学与计算机模拟相结合的技术,基于石油各馏分性质与其化学分子组成息息相关的理论,根据石油馏分有限的分析数据,构建虚拟分子集,该分子集的组成与结构信息由合适的计算机方法表示,在不损失关键组分和结构信息的前提下,尽可能压缩分子集的规模,以便于过程模型开发和物性预测[5]。整个建模过程关键在于3个方面:一是选择表示石油中大量复杂分子的方法;二是准确计算各类石油分子的物性,以及选取合理的混合规则确定石油馏分的宏观物性;三是以已有的分析数据作为目标函数,通过优化相关参数,对石油分子组成进行合理和准确的定量计算,实现从石油馏分物性到分子组成的转变。
1 石油分子重构技术
石油分子重构技术大致可以分为2部分:石油分子的表示方法和石油分子组成的构建方法。石油分子的表示方法主要有:键电矩阵法、结构导向集总法、结构单元耦合原子拓扑矩阵、分子同系物矩阵法、伪化合物矩阵法、状态空间表示法。石油分子组成的构建方法主要有:随机重构法、最大信息熵法、随机重构-最大信息熵法。
1.1 键电矩阵法
键电矩阵是基于有机化学反应本质上是官能团反应提出的,而官能团发生的反应大多可从文献上查到。Core[6]首先提出了利用计算机来辅助构造复杂化合物的路线,将化合物分子以二进制的方式存储于计算机中,并为化合物的反应路径制定了编写规则。Ugi等[7-9]认为分子可以由原子、化学键、电子组成,由此提出了键电矩阵,而化学反应可由反应矩阵表示,键电矩阵和反应矩阵可以作为编写计算机程序的基础。Horton等[10]将该方法用到石油分子的表示中,并定义了反应矩阵表示分子的操作,官能团拼接由矩阵加法完成,断键操作为-1,成键操作为+1,其具体形式如图1所示。键电矩阵法将各种化合物存储于计算机中,通过合理编写反应路径,使化合物合成路径的网络构建以及不同化合物之间的化学反应预测成为可能。
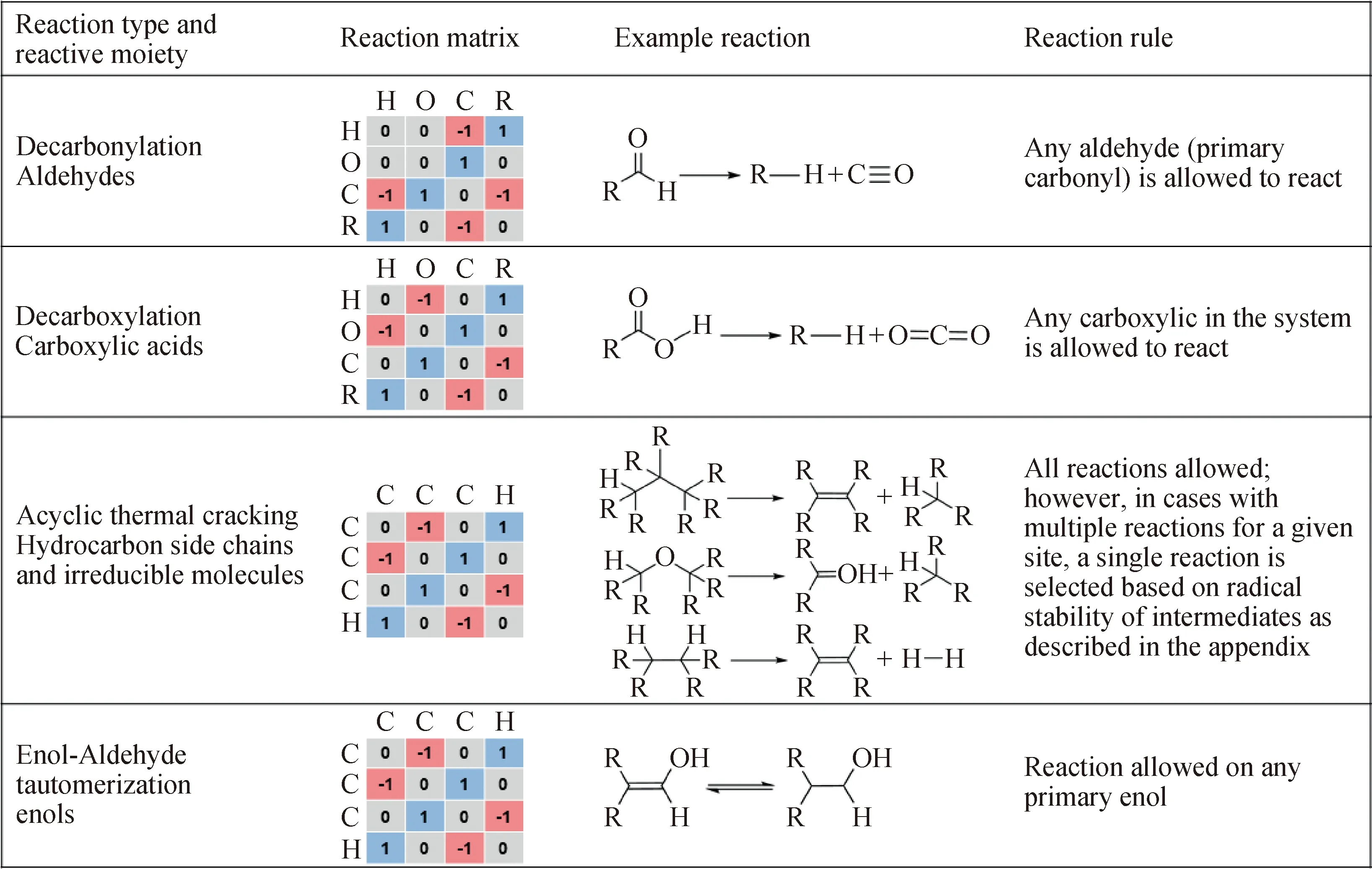
图1 分子反应矩阵[10]Fig.1 Molecular reaction matrix[10]
1.2 结构导向集总法
结构导向集总(SOL)法最早由Quann和Jaffe[11]于1992年提出,即用少部分简单的结构单元来表示化合物的组成。他们引用基团的概念,选取22种结构基团来描述化合物的分子结构,用这些结构基团的组合向量来表示石油分子。其中,包括3个芳烃结构基团(A6、A4、A2)、6个环烷结构基团(N6、N5、N4、N3、N2、N1)、1个—CH2—侧链(R)、2个异构侧链支点(br)和环上的甲基(me)、1个代表不饱和度的氢基团(IH)、1个联苯间的桥基团(AA);另外还有2个硫基团(NS、RS)、3个氮基团(AN、NN、RN)、3个氧基团(NO、RO、KO)。结构基团如图2所示。

图2 结构基团示意图[11]Fig.2 Schematic diagram of structural groups[11]
为了将结构基团表示化合物的方法用于油品的宏观物性预测,Ghosh和Jeffe等[12]首先利用SOL法的思想,将气相色谱法得到的57个碳氢化合物结构基团与汽油辛烷值关联起来,建立了汽油辛烷值预测的通用模型。随后,Ghosh等[13]根据相同的思路,建立了柴油十六烷值预测的通用模型。SOL法也适用于分子动力学模型,纪晔等[14]提出了一种基于结构导向集总的催化重整过程建模方法,能够从分子水平描述催化重整装置原料分子到产物分子的转化过程。该方法使用352种原料分子,17条反应规则,建立反应网络,包含中间产物和产品共533种分子,共有分子反应转化路径617条。对于不同的工况,产品分子组成的模型预测,其平均相对误差均小于3%。Nguyen等[15]建立了基于SOL法的轻瓦斯油加氢脱硫(HDS)的集总动力学模型。该模型包含16个结构基团,以这些基团为基础建立的模型,能预测出反应温度范围为200~375 ℃的轻瓦斯油加氢脱硫反应从原料输入到产品输出过程的分子结构类型的变化。最初的SOL法主要用于表示轻油的分子结构。Kumar等[16]针对VGO(减压蜡油)的分子组成特点,按其特征分为16个基本结构,在此基础上,将带有甲基侧链的异构体划分为45个亚类,基于化学反应动力学方法,建立了加氢裂化分子反应动力学模型。
为了使该方法推广到渣油分子结构的表示中,Jaffe等[17]在原有22个结构基团的基础上,对沥青质的群岛结构基团进行了大量的扩展,加入了Ni、V等结构基团,将其应用到了减压渣油分子的表示中。倪腾亚等[18]采用SOL法对渣油馏分进行分子重构,设计了21个结构单元来表示烃类结构、杂原子结构及重金属结构,用55类结构向量来表示2791种典型的渣油分子。采用模拟退火算法计算渣油的分子组成,使烃类组成信息和平均分子结构参数的计算值与分析测定值差异最小,构建了SOL法的渣油分子组成计算模型。模拟结果表明,渣油的残炭、密度等性质参数,以及芳碳率、芳环数等结构参数的计算值与实验测定值吻合较好。Tian等[19]以SOL结构向量为基础,假设渣油分子由92种单核种子分子和46种多核种子分子组成,以7004个结构基团表示各种不同的渣油分子,构建了延迟焦化产品分布预测模型,模拟结果表明,产品液体收率与反应温度、原料的H/C比和循环油量与新鲜原料油量的比值有关。
Yang等[20]针对传统集总法的不足,提出了一种基于结构导向集总(SOL)法和蒙特卡罗(MC)相结合的方法来模拟催化裂化(FCC)汽油的二次反应过程。他们采用SOL法来表示原料和产品的分子结构,并建立了60余项反应规律,得到了FCC汽油的所有二次反应网络。又由蒙特卡罗积分法计算各分子的反应概率,得到产物的分布。以中国工业催化裂化装置的3个催化裂化汽油样品为模拟对象,验证了所提方法的有效性。
He等[21]为了解决SOL法机理模型复杂度高、计算效率低的缺点,提出了一种混合建模的方法。即采用CatBoost算法构建数据模型,弥补简化SOL机理模型造成的精度损失,结合机理模型,既保证了预测的准确性,又大大缩短了计算时间。对某MIP装置产物的产量和产率进行了预测,结果表明,模型在保证预测精度的同时,其计算时间大幅减小。
邱彤等[22]为了实现石油炼制全过程的分子管理,基于SOL法中对分子的表示方法,提出了一种新的石油馏分分子重构模型。该模型选取22个结构特征参数来表示石油馏分,通过蒙特卡罗模拟产生虚拟分子,最后用最大信息熵的思想优化调整虚拟分子的含量,建立的模型既能在宏观上预测石油馏分的物性,又能在微观上确定石油馏分的分子含量。
虽然用结构基团可以直接计算各分子的元素组成和相对分子质量,从新的结构基团出发也可关联计算出各分子的沸点和相对密度等物性参数,但随着分子尺寸的增大,这种方法不再使表示分子的结构向量与该分子一一对应,为此研究者提出了一些约定条件,简化构建分子的复杂性,取得了不错的效果。总体来说,SOL法较好地表示了石油分子的结构特征,而且本身具备扩展性,是一种较为合理的分子重构方法。
1.3 结构单元耦合原子拓扑矩阵
石油中较重的分子往往含有一些杂原子,这些杂原子与复杂的核心原子和侧链结构相连,形成杂原子结构向量及双键的位置可能各不相同,使传统的结构导向集总法不能明确表示。为了减少分子物性数据计算及反应位点的识别错误,建立结构单元与键电矩阵的准确映射,使石油分子结构的生成更加方便,中国石油大学(北京)分子管理课题组结合键电矩阵和结构导向集总的特点,开发出了结构单元耦合原子拓扑矩阵。该课题组Chen等[23]对SOL法进行扩展,并将扩展后的结构单元分为核心结构单元和侧链结构单元2类,这些结构单元被称为CUP结构单元,如图3所示。该表示方法被用于建立催化裂化油浆在滴流床加氢处理装置中的分子反应动力学模型,模型包含5753个分子和15830个反应网络,模拟后的结果表明,产品性质的预测结果与实际值吻合较好。随后,该课题组Liu等[24]选择CUP结构单元中的部分结构,建立了汽油分子结构与其辛烷值的关联模型。Cai等[25]以CUP结构单元为基础选出15个结构基团、20个联合基团以及相对分子质量作为化学特征量与烃类化合物的黏度进行关联,基于人工神经网络,建立了定量结构-性质的关联模型,该模型能准确预测纯碳氢化合物在不同温度下的黏度。

A6—A six carbon aromatic ring; A4—A four carbon aromatic ring; A2—A two carbon aromatic ring;1,2 represents the number of structure group.图3 CUP结构单元划分和实例[23]Fig.3 Division of CUP structural units and instance[23](a) Core structural unit; (b) Side face structural unit; (c) CUP structure unit representation ofphenol containing 4-ring aromatic ring and its combination logic
在键电矩阵的基础上,结构单元原子耦合拓扑矩阵实现了CUP结构单元与键电矩阵的映射,有利于分子结构的绘图,并获得分子的通用化学标记语言和通用分子存储格式文件[26],以芘分子的生成为例,如图4所示。
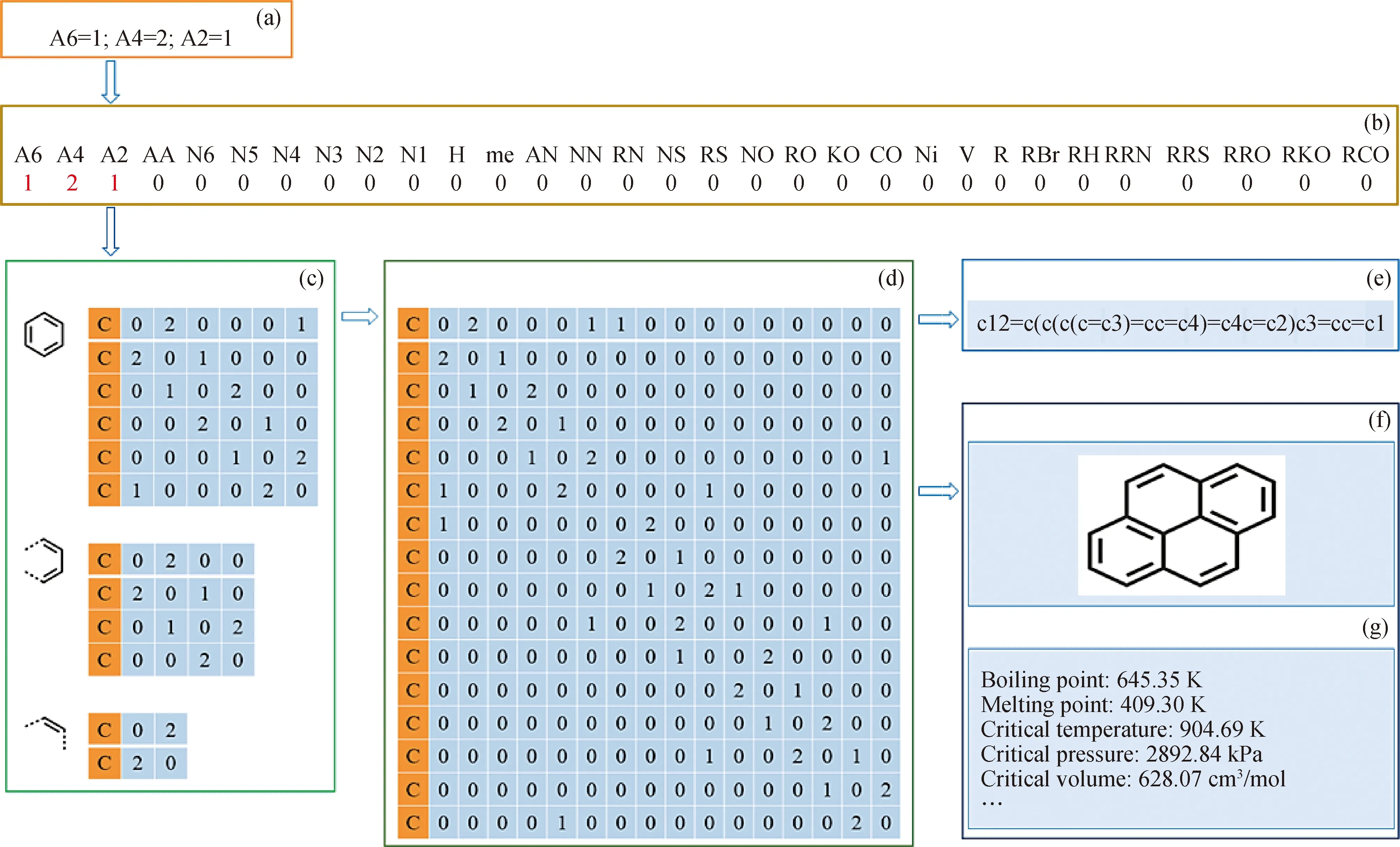
图4 芘单分子的多层次构建流程[26]Fig.4 Multilevel construction process of pyrene monolecule[26](a) Structure unit string; (b) Structural element vector; (c) The bond electric matrix of different elements;(d) Atomic connection information table; (e) Eommon chemical markup language information code;(f) Image generation; (g) Calculation of physical and properties
结构单元原子耦合拓扑矩阵不仅有利于调用分子物性预测模块进行分子的物理化学性质预测,而且容易将分子连接到分子模拟或流程模拟软件中,是一种较为合理的石油分子表示法。
1.4 分子同系物矩阵法
1999年,英国曼切斯特大学的Peng[27]提出了用分子同系物矩阵(MTHS)法来表示石油馏分的方法。该方法中,矩阵的行数代表该类型分子的总碳数,矩阵的列代表不同的分子类型,行与列的交叉处代表该类型碳数的分子占总分子的质量/摩尔分数,同种类型的分子碳数分布符合Gamma分布。如图5所示。
MTHS法有2个应用步骤:一是模型分子信息矩阵的建立,根据不同石油馏分所对应的碳数分布范围和所含分子结构特点确定;二是物性数据的传递和优化。将MTHS中烃类分子的物性数据,通过混合规则计算出石油馏分的宏观物性,与对应的实际石油馏分分析数据进行比较,根据优化算法确定MTHS分子组成[28],如图6所示。MTHS法在实际应用中,既能通过石油馏分的宏观物性确定其分子组成,又能由确定的分子组成预测石油馏分的宏观物性。
Zhang[29]基于Peng[27]的研究工作,将石油馏分的碳数与相对分子质量、沸点和密度相关联,其他物性数据通过经验关联式由沸点和密度数据计算得到。然而,石油馏分宏观物性的预测是基于简化后的分子类型进行的,没有考虑每个矩阵同系物族内同分异构体的贡献。且由于基本假设过多,而忽视了分子结构对其宏观物性的影响,例如,辛烷值、蒸气压和黏度等。因此,有必要在每个矩阵的内部对同分异构体进行划分。Aye等[30]充分考虑到同分异构体结构对馏分宏观物性的影响,通过设置集总规则,对同分异构体结构进行了归类和简化,然后利用插值法计算MTHS中馏分的分子组成,但该方法提出的结构-性质关联式只限于轻馏分。Ranzi[31]研究发现,同分异构体矩阵内部的热力学平衡是固定的,该规律可以用MTHS法来计算石油馏分的宏观物性。除了同分异构体的影响,作者还创建自动选择样本矩阵的转换方法,通过线性组合生成新矩阵,新矩阵的数学模型由1个目标函数和几个约束条件组成,其作用是使MTHS法所计算出来的物性预测值与实际测量值之间的误差最小。在计算石脑油宏观物性时,由于物性关联公式和混合规则均存在一定程度的误差,使MTHS法计算出来的物性数据偏离真实石脑油物性数据的可能性变大。为了解决上述问题,白媛媛等[32]在预测石脑油宏观物性时,以简化后的MHTS表示石脑油分子组成,同时根据石脑油样本的特点,对决策变量加入一些有效的约束,进一步提高模型预测的准确性。以往MTHS法的研究仅限于汽油等轻质石油馏分,并且只考虑了碳氢分子。为了将其应用扩展到中、重质石油馏分中,Ahmad等[33]利用基团贡献法,根据石油中化合物的分子结构,通过最小化模拟值和实测值之间的差异来生成石油馏分的分子矩阵,并由MTHS法计算石油馏分中单个分子的宏观物性;除此之外,还在MTHS的列中引入含硫和含氮同系物结构,对含硫和含氮化合物的物理化学性质和热力学性质进行预测,使该方法能更好地应用到重油中。
随着石油馏分变重,碳数分子规模增大,不可避免地导致同分异构体分子的数量大幅度增加,此时确定石油馏分MTHS中的分子组成将变得十分困难。为了解决上述问题,Gomez-Prado等[34]首次提出利用石油馏分沸点切割代替碳数划分。郭广娟等[35]根据柴油分子组成特点,以柴油沸点代替碳数划分,建立MTHS法模型,分别对催化柴油、加氢改质柴油、加氢裂化柴油进行烃类组成预测,结果吻合较好。李洋等[36]利用MTHS法对直馏蜡油、焦化蜡油、催化蜡油进行了分子重构,得到不同烃类结构在各馏分段的分布,模型预测值与实测值吻合较好。侯栓弟等[37]针对环烷烃、芳烃结构协同关联关系,考虑不同化学结构对蜡油烃类宏观物性的影响,提出了27个虚拟官能团的分子重构模型。对于不同的原料而言,同一沸程对应的分子组成并不相同,由此建立的模型可能存在多解,导致了模型的泛化能力受到限制。Liu等[38]改变了MTHS的表示方法和变换方法,在碳数行中嵌入伪组分及其化学信息,使得改进后的MTHS很容易地扩展到较重的组分,在保证精度的前提下,极大地提高了模型的可用性。Wu等[28]利用改进的MTHS法对汽油和柴油进行分子组成模拟,在计算模型的分子组成时引入了分布函数的思想,即同类型的分子含量服从Gamma分布的规律,这样既减少了多解带来的不确定性,又增加了计算结果的准确性。Wang等[39]提出了表征石脑油馏分分子矩阵的新方法,MTHS法中的虚拟分子由碳数相同的真实分子的同分异构体集总获得,并用正态分布代替Gamma分布来描述分子矩阵中分子分布,结果表明,这种新型分子矩阵能够表示更详细的石脑油分子信息,并以较少的未知参数提高模型预测石脑油馏分宏观物性的精度。
根据石油不同馏分的碳数分布范围确定适宜的MTHS大小具有重要的意义。Hu等[40]以MTHS为基础,建立了一种油品宏观物性与分子组成互相计算的方法,针对不同的石油馏分,选择合适碳数分布的MTHS,然后,利用这些分子信息来建立分子模型。这些模型被纳入到炼油厂各装置的整体优化中,可以用来选择合适的原料、产品以及优化工艺操作条件。
MTHS法以常规分析得到的纯物质宏观物性数据库和以矩阵表示的石油分子库为基础,用石油馏分的分析数据为预测目标,根据混合规则,选取适当的优化算法来确定石油馏分的分子组成。总体而言,MTHS法直观地表示了石油馏分,对轻油的处理较为成功。
1.5 伪化合物矩阵法
伪化合物矩阵(Pseudo-Compounds Matrix)法由法国石油研究院(IFP)开发,构建方式与分子类型同系物矩阵(MTHS)法相反,矩阵的不同行代表不同的同系物类型,列代表碳数,但划分的同系物结构更复杂,将烷烃、芳烃等划分为一环、二环、三环、四环等结构。由于其本质是根据石油馏分的特点选取有代表性的分子建立石油分子库,所以将与伪化合物矩阵相似的建模方式一起讨论。
Hudebine等[41]对比各种分子重构方法之后,开发出伪化合物矩阵,对直馏柴油、轻循环油和焦化汽油的质谱、硫形态、氮分析、密度和模拟蒸馏5个方面进行表征,构造了612种伪化合物,覆盖28个不同的族组分,在预测黏度、十六烷值、氢含量、芳烃含量等宏观性质方面取得了较好的精度。
Ramirez等[42]针对柴油加氢裂化装置,选用40个分子代表原料柴油的组成,113个分子代表柴油加氢裂化产物的组成,并将这些分子导入到稳态过程仿真器(Pro/Ⅱ-Simsci)中,建立了柴油加氢裂化装置的预测模型。对应的产品产量和其宏观物性预测结果表明,预测值与实验值吻合较好。随后,他们采用由傅里叶变换离子回旋共振质谱联用正电喷雾电离源、负电喷雾电离源以及常规标准化分析方法获得的信息,对减压渣油和常压渣油进行分子水平表征,每个渣油分子由150个结构特征量表示,将渣油的整体分子结构特征和宏观物性一一对应起来,建立了渣油宏观物性的预测模型,以新的渣油样品验证了该方法的有效性[43]。
Albahri[44]利用68种纯组分建立预测模型,模拟了石脑油在精馏塔上的分离过程。该模型比目前使用的表征方法具有更高的准确性。该方法操作灵活,可根据需要调整任一纯组分,并可进一步用于石油馏分宏观物性的预测。
崔晨等[45]对汽油分子进行适当的简化,用166个分子构建了汽油分子库,并采用模拟退火算法进行优化,通过汽油的宏观物性反算其分子组成。采用一套催化裂化汽油的数据对模型进行了验证,所得汽油宏观物性的预测值与实验值平均相对偏差小于2%,所有宏观物性数据的平均相对偏差小于5%。
与其他重构方法相比,伪化合物矩阵法建模速度更快更精确。但其需要通过模拟蒸馏、质谱、硫化学发光气相色谱分析检测和氮分析等分析方法给出更多的信息;同时,受到核结构数量的限制,故其主要是针对轻、中质石油馏分的分子重构。
1.6 状态空间表示法
最近,Mei等[46]提出了新概念——状态空间表示法。该方法根据石脑油馏分的特点,将其分为多种不同的同系物。1个系列的同系物用1个向量单元表示,所有的向量单元一起组成多维非负欧几里得状态空间。因此,任意1种石脑油都可以表示为状态空间中的1个点。Mei等认为由同系物组成的向量单元两两正交,因此,任意石油馏分在状态空间中都可以表示成不同向量单元的线性组合。非负矩阵分解(NMF)算法[47-49]利用向量单元预测样品组成并消除冗余信息和测量误差。这种表征方法减少了石油分子库的单元数量,剩余的向量单元即构成状态空间。与传统方法相比,石脑油数据库中向量单元的数量明显减少,降低了计算成本,更适合实时控制和优化。然而,状态空间表示法很大程度上取决于向量单元选取的完整性,也需要大量的实验分析。目前,只应用于对石脑油的分子重构,其普适性还有待验证。
2 石油分子组成的构建方法
2.1 随机重构法
随机重构(SR)法的提出,基于石油分子基团分布规律的认识,考虑到这些分子基团的分布规律符合概率密度分布,可以通过随机抽样选出大量的分子基团来组成虚拟分子库,并用这些虚拟分子库来表示实际的石油馏分。该方法主要包含4个关键的步骤:(1)在计算机上生成一系列的结构基团框架。(2)引入概率密度分布函数来表示这些结构基团在石油中的分布。(3)以石油馏分的各种宏观物性为目标,利用蒙特卡罗法对大量虚拟的基团进行多次随机抽样,如图7所示,优化概率密度分布函数的参数,确定合适的虚拟分子库。(4)将抽样所得分子库的宏观物性数据与目标分析数据相比较,计算其数学期望、标准偏差等统计特征量,得到所求问题的近似解。
1990年,Neurock等[50]提出用SR法来模拟重油的分子组成。随后,其所在的Klein组的研究人员[51-53]开展了一系列工作,将SR法用于石油各馏分的分子重构中,通过构建几万到几十万个虚拟分子,使石油馏分各物性概率分布函数参数保持稳定。但是,大规模虚拟分子的构建不利于当时计算机的处理。1997年Campbell等[54]根据石油馏分的分析数据,使用蒙特卡罗抽样法对渣油中的基团分布进行抽样,优化概率密度函数的参数,确定出原料分子库,此时,对分子库的宏观物性进行核算,若模拟所得分子库的宏观物性和渣油分析数据差距满足要求,则保留;若不满足,则通过全局优化算法对分布函数进行优化,直至分子库的宏观物性达到要求,整个过程如图8所示。若抽样后所得分子数量过大,则根据Campbell的积分法,将分子数压缩到几百个,比较预测的性质数据与实验值的差异,调整其分子的质量分数,直至获得最优分子的最优质量分数。

RN—Random number (0—1.0)图7 蒙特-卡罗抽样过程[50]Fig.7 The process of Monte-Carlo sampling[50]
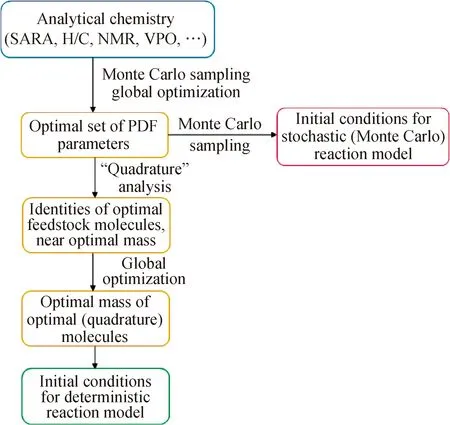
SARA—Saturate, aromatic, resin, asphaltene;H/C—Hydrogen/Carbon ratio;NMR—Nuclear magnetic resonance;VPO—Vapor-pressure osmometry;PDF—Probability density function图8 随机重构法构建虚拟分子库[54]Fig.8 Stochastic reconstruction method toconstruct virtual molecular set[54]
Trauth等[55]对渣油体系进行了细化,按四组分进一步划分,根据各组分的分布规律,选取相应的分布函数类型,如图9所示。随后,Verstraete等[56]对Trauth等的工作做了进一步的补充:以渣油为研究对象,除了划分为四组分外,还引入了硫化物和氮化物。之后这套方法被不断完善,形成了一套完整的计算体系,与分子动力学软件KMT和KME整合到一起,实现了从原料到产物的性质预测[57]。

A—Core structure and additional side chains图9 随机重构构建渣油分子库方法[55]Fig.9 Residue molecular set constructed by random reconstruction[55]
虽然经典的SR法在轻、中质石油馏分的应用中能够产生合理的分子库,但随着馏分的变重,要得到精确的分子库变得愈发困难。Deniz等[58]在传统的SR法基础上,引入了扩展的参数集,并将其应用在6种不同沥青质分子的重构中。相比传统的SR法,扩展的参数集使沥青质分子重构模型的预测值与实际值的误差下降45%~85%,并且在不牺牲生成分子组成空间的情况下提高了SR法的拟合能力。为了减少重构分子所需的时间,Deniz等[59]提出了一种基于人工神经网络和遗传算法相结合的石油馏分随机重构方法。首先,利用人工神经网络合理的确定了优化空间中初始结构参数集。然后,利用遗传算法对初始参数集进行优化。模拟结果表明,这种混合方法在保持准确性的前提下,大幅地减少了优化参数集所需的时间。
针对重质石油馏分,要得到更为精确的虚拟分子库,概率密度函数的分布参数的优化起着至关重要的作用。经典的优化方法需要合理确定石油馏分中分子结构的概率密度分布函数,且极易陷入局部极小值,影响了SR法的准确性,为此,一些研究者选择随机优化算法来解决上述问题。Glazov等[60]引入了一种算法,该算法可以根据一些重质石油馏分的统计检验分析数据确定哪种分析指标影响结构属性的分布。其核心思想是根据分析数据和分布参数之间的显著相关性,优化分布参数的取值。结果表明:不同类型的分析数据会影响重构馏分的不同参数,蒸馏曲线数据是方差分布信息的主要来源;元素分析和PNA有助于确定各种结构属性的平均值;分子中支链数量的信息与13C NMR数据紧密相关。Ashraf等[61]开发了一种软件来求解模型分布参数,使用了蒙特卡罗(MC)、最大似然估计(MLE)、马尔可夫链蒙特卡罗(MCMC)、动态维数搜索算法(DDS)等10种随机方法确定出重要的模型分布参数。结果表明,所有算法中,最大似然估计算法和动态维度搜索算法提供的参数更为可靠。
SR法通过随机抽取一套虚拟分子来表示石油馏分,当抽取的分子数量足够大时,可以更好地反映石油分子的组成特点。但该方法依然存在不足,一方面,随机抽取的方式可能会组合出石油中不存在的分子;另一方面,SR法的算法执行速度较慢,对于重油馏分尤其如此。
2.2 最大信息熵重构法
最大信息熵(EM)最早由Shannon等[62]于1948年提出,其理论本质是根据部分分布确定信息对未知分布不确定的信息进行最合理的推断。2004年,法国石油研究院的Hudebine等[63]将其引入轻馏分的分子重构中,形成了REM法,作为两步法(随机重构-最大信息熵)的第二步(详见2.3节随机重构-最大信息熵法),基于最大信息熵的目标函数表达式如下所示。
(1)
(2)
(3)
(4)

该方法以信息熵最大化为目标,根据油品的分析数据及其分子的混合规则,调整优化相关参数,从已经预设好的石油分子库中确定各分子含量,具体过程如图10所示。随后,他们进行了一系列改进[64-67],将该方法应用到重馏分的分子重构中。

图10 最大信息熵法流程[63]Fig.10 The process of the Entropy Maximization Method[63]
顾江华等[68]使用REM法对石脑油馏分进行了分子重构。研究中选用石脑油馏分密度、馏程数据以及详细烃族组成数据作为石脑油样品的物性数据,同时选用合适的方法对预定义的石脑油分子库中分子物性进行计算,最后使用REM法确定石脑油馏分的分子含量。模拟结果表明,重构出来的混合分子的物性与石脑油馏分的物性相对误差较小。Alvarez-Majmutov等[69]将先进的建模技术与常规的石油表征方法相结合,重构出中间馏分的分子组成。
该方法将油品分析表征数据作为输入,根据一套化学/热力学标准,构建出5000个代表性的虚拟分子。利用蒙特卡罗抽样法抽取分子,并根据最大信息熵原理优化了各分子的摩尔分数。2个具有代表性的中间馏分样品的模拟结果表明,模拟出来的中间馏分的物性和烃类组成,与对应样品的物性分析数据和烃类组成非常接近。Pan等[70]基于最大信息熵理论,结合结构导向集总(SOL)和蒙特卡罗(MC)方法,在分子水平上模拟了催化裂化(FCC)装置的反应原料,结构导向集总(SOL)法用于表示原料分子结构,蒙特卡罗法用于生成一个预定义的分子库,最大信息熵法用于确定分子含量,对原料性质的模拟结果中,烷烃、烯烃、环烷烃、芳烃等主要组分的平均相对分子质量和质量分数与对应样品实验数据吻合较好。硫化物是石油产品中需要控制的重要指标,由于其结构复杂,不宜对其进行分子重构,Bojkovic等[71]基于REM法,依据有限的分析数据,重构出了常压蜡油和减压蜡油中的硫化物分子。需要指出的是,只有在传统分析指标(如沸点、密度、PINA质量分数、总硫量和芳香硫量)的基础上,才能准确地重构出含硫化合物的详细分子组成。牛莉莉[72]将REM法引入对大庆原油的分子重构中,模拟结果表明,原油的碳含量、氢含量、平均分子质量、硫含量、氮含量、窄馏分收率、各组分族组成的模拟值与实验值基本一致。
石油分子重构技术中,想要得到较为准确的分子含量,参数的优化往往是其关键一环。在REM法中,一般采用拉格朗日乘子的形式,将混合规则转化为线性约束,建立满足这些线性约束条件的最大熵判据,然后采用共轭梯度法对熵判据进行优化,以确定分子库中每个分子的摩尔分数。Pyl等[73]比较了最大信息熵、人工神经网络和多元线性回归3种方法对于石脑油的分子重构,REM法在有足够的石脑油蒸馏数据提供碳数分布信息的前提下,不仅石脑油物性的模拟精度媲美人工神经网络,而且不易受石脑油种类变化的影响。传统的优化算法比较经典,但训练效率低,且容易收敛到离初始点最近的局部解。近年来,随着各种算法逐渐被开发出来,参数的优化算法有了更多的选择。Chen等[74]开发了一个三步重建算法,根据部分分析数据重构出一组代表性的分子。首先,对石油馏分的结构分布参数进行优化,然后利用优化后的参数确定石油馏分分子库,最后采用REM法得到分子含量。Bi等[75]提出了一种新的分子重构方法。首先,利用自适应云模型构建了由伽马分布趋势、区域特征、权重特征和不确定性组成的有效概率密度函数,然后使用一种遗传算法-粒子群算法组合的优化方法对概率密度函数的参数进行优化,确定出初始的虚拟分子库,与其他方法相比,该新的优化方法重构出来的分子更接近实际组成。随后,Bi等[76]采用遗传算法-粒子群组合算法(GA-PSO),对用于原油性质预测的支持向量机(SVM)模型进行优化。优化过程和结果表明,新提出的GA-PSO-SVM方法比经典的GA或PSO方法更省时、更准确。与经典的网格搜索支持向量机模型进行比较,组合GA-PSO-SVM模型更加适合用于原油性质预测任务。
REM法根据石油馏分的分析数据,不断调整预设好的分子库中各分子的含量,使其模拟值和实验值相吻合。这种方法强烈地依赖于预设的分子库。如果初始分子库的物性数据与所测石油馏分的分析数据差距大,求解过程就会变得困难。该方法的优点是计算迅速,缺点是前期需要做大量的分析工作。
2.3 随机重构-最大信息熵法
为了减轻SR法的计算负担,Hudebine等[63]提出了一种结合随机重构和最大信息熵的建模方法,这种新方法称为随机重构-最大信息熵(SR-EM)法。在此两步法中,第一步是通过SR法生成等摩尔混合物作为初始分子库。第二步,将限定的线性约束条件调整到最大熵判据的公式中,构建最大熵判据。选取相应的算法并调整初始分子的摩尔分数,使与约束相关的信息熵最大化,求得熵判据的最大值所对应的分子摩尔分数即为最终结果,SR-EM法的流程图如图11所示。Hudebine等[63]将SR-EM法应用到轻循环油(LCO)分子重构中。首先,通过SR法获得5000个等摩尔分数的分子;然后,在第一步得到的5000个初始分子库的基础上,结合LCO的分析数据,使用EM法确定最终代表分子的摩尔分数。

SR—Stochastic reconstruction; EM—Entropy maximization图11 随机重构-最大信息熵法流程[63]Fig.11 Flow diagram of the SR-EM method[63]
Verstraete等[64]用两步法实现了减压蜡油的分子重构,模拟得到的混合分子的物性与真实样品的物性非常接近。其后,Verstraete等[56]将SR-EM方法扩展到渣油馏分的分子重构中,设计了适合渣油馏分结构属性的分布图和结构图,不仅能表示渣油馏分中的碳化物、氢化物、硫化物结构,而且能表示氧化物和氮化物结构。Oliveira等[77]构建了两步程序来模拟LCO柴油加氢处理过程, SR-EM法被用于表示第一步的原料,随后,对SR-EM法进行扩展,使其应用到减压渣油中。首先,通过SR法获得一组预定义的分子库;其次,通过EM法得到分子库中分子的摩尔分数。SR-EM法的有效性在8个减压渣油样本中得到了验证[78]。紧接着,Oliveira等将SR-EM法应用于减压渣油加氢处理动力学模型中,利用重构出来的分子库表示原料,从而克服了减压渣油分子细节的缺乏[79]。Alvarez-Majmutov等[80]将传统的SR-EM法与先进的石油表征方法(GC×GC-FID,GC×GC-SCD)结合起来,在分子水平上获得中间馏分和减压馏分的分子组成,并进一步开发出减压蜡油加氢裂化分子水平的结构-反应模型[69,81]。
尽管SR法和REM法都可以作为独立的方法来构建石油分子,但是每种方法都有其局限性。SR-EM方法克服了这2种方法的缺点。单独使用SR法时,会将分子结构基团的重复抽样和迭代放置在优化循环中,这使SR法的计算量很大。单独使用REM法时,计算量要小得多,但是,这种方法在很大程度上依赖预设好的分子库,如果初始分子库的物性数据与实际样品的分析数据相差较大,则容易导致优化后计算出来的个别分子摩尔分数很大,而大部分分子的摩尔分数为零。SR-EM方法不仅避免了SR法对分子结构基团的重复抽样,而且还为REM法提供了可靠的分子库,既大大减轻了计算负担,又保持了良好的准确性。
虽然不同的石油分子重构方法的表现形式、建模过程和解决方法可能各不相同,但每种方法基本上都要经过6个步骤:分子库的建立;获取纯组分的物性数据;混合物宏观物性的计算;建立目标函数;优化相关参数;调整分子的摩尔分数。图12为整个石油分子重构步骤的流程示意图。

图12 石油分子重构方法[82]Fig.12 The petroleum molecular reconstruction method [82]
混合规则被用来计算混合物的平均宏观物性,石油馏分表征获得的物性数据用于建立目标函数。此外,还需要选取相应的算法,优化相关参数,调整分子库中分子的摩尔分数,使得石油馏分的模拟值与目标值的差异最小。目前,各种石油分子重构方法已被成功地应用于石油加工过程中,但仍有一定的改进空间。纵观石油分子重构方法的发展历程,早期的文献主要关注模型对石油馏分宏观物性的预测误差,近期的文献更加关注石油馏分分子组成的模拟精度。值得注意的是,预测石油馏分的宏观物性往往比预测分子组成的准确性好。
3 石油分子重构各方法的特点分析和比较
总体而言,石油分子重构技术主要分为石油分子的表示方法和石油分子组成构建方法2个过程。其中,键电矩阵法通过矩阵来表示原子的连接方式,比较简单直观,但是随着相对分子质量的增大,所需矩阵的维数也随之增大。尤其对于重油组分,大的分子数目和相对分子质量会极大地消耗计算机的内存。此外,对于多环化合物,环与环之间重合连接处、原子与原子连接处的表示不够直观,也为石油分子的编写带来许多麻烦。但其优点是计算机对矩阵表示的分子结构容易处理,并且执行时速度较快。目前,许多现代化学信息学软件管理系统分子的构建方式依然采取键电矩阵法。
结构导向集总(SOL)法的核心思想是:石油分子由有限的结构向量组成,通过结构向量加上侧链的形式来表示石油分子,既简单又直观,同时也方便石油分子物性的计算。但是随着分子尺度的逐渐变大,该方法的分子向量与分子不再一一对应,而是一个分子向量对应多个分子。此时,由物性分析数据选出来的分子存在多解,导致相同的宏观物性解出的石油分子差别较大,影响了石油分子计算的重复性和准确率。另一方面,对于分子反应动力学模型而言,用结构导向集总来表示分子反应是一种有效的方法,但由于整个反应部分相当于结构基团的拼接和组合,所以该方法只能做到反应路径层面,难以深入到机理层面。
同系物矩阵(MTHS)法以矩阵的行代表碳数,列代表同系物族,直观地将石油分子表示出来。除了有利于石油分子在计算机中的存储外,还可以通过改进将同分异构体的宏观物性差异考虑进去,使得重构出来的分子更加符合实际油品组成。另一方面,MTHS法基于预设好的石油分子库和相应的物性数据,对于选取的石油分子含量的计算更为合理。不过,预设石油分子库受到分析水平的限制,比柴油更重的油品分子可能并不存在于石油分子库中,这也使得基于MTHS法重构出来的分子并不准确。除此之外,为了使重构出来的石油分子更加符合实际,石油分子库的建立需要收集各类不同地区的油品并对其进行详细的分子组成分析,前期工作量较大。而且由于组成油品分子的数目巨大,有些石油分子的宏观物性获取也极为困难,这也给MTHS法的应用带来不便。总体而言,MTHS法对于柴油馏分以下的油品,其重构出来的分子较为准确。但受限于目前计算机存储能力和计算能力以及分析水平,MTHS法不容易应用到重油的分子重构中。
随机重构(SR)法基于石油分子呈现一定的概率密度分布,依靠蒙特卡罗抽样法,将不同的基团以一定的先后顺序确定下来,并采用特定的方式进行拼接。每抽取一组结构基团就能确定一个石油分子,每次模拟过程确定一个石油分子库,这种方式的好处是,通过确定石油分子中结构基团的分布规律即概率密度分布函数(PDF),进而确定石油分子的分布规律,比较契合实际油品中分子的分布状况,而且该方法将数量巨大的石油分子进行适当的简化,减少了变量个数,为其石油分子含量的计算提供了便利。目前SR法被广泛应用于石油的全馏分分子重构,是一种有效的模拟方法。但该方法除了利用一些算法在计算机中将各种结构基团选取出来,还要逐步确定不同基团之间的拼接重组,进而形成石油分子,使整个过程变得相对复杂。另一方面,抽样过程中可能生成石油中并不存在的分子,这将导致由SR法模拟出来的宏观物性的计算结果虽然与实际油品宏观物性一致,但分子组成并不相同,影响了结果的准确性。
最大信息熵(REM)法提供了一种计算石油分子含量的方法,以石油馏分有限的分析数据为约束条件,根据混合规则,优化相关参数,调整石油分子库中的分子含量,在保证信息熵最大的同时,使石油的宏观物性模拟值与实验值之间的差异最小。依据最大熵判据,该方法除了可以调整石油分子的摩尔分数外,还能剔除不符合要求的分子。但是,REM法的应用除了需要较为完备的分析数据外,还需要输入确定的分子结构信息,保证计算结果的准确性。
随机重构-最大信息熵(SR-EM)法结合了SR法和REM法各自的优点。SR法根据石油分子结构基团的分布特点生成一套初始分子库,较为合理;但后续详细分子含量的计算比较复杂且速度较慢;而REM法比较依赖于合理的初始分子库,计算分子含量时比随机重构法的速度快很多,且准确性更高。实际应用时,相比单独的SR法或REM法,SR-EM 法进一步提高了模型的准确性和适用性。
石油分子重构技术是石油炼化分子管理的基础,其根据石油馏分分子的分布和结构特点,结合计算机模拟方法,将有限的分析数据转化成详细的分子信息。这些分子信息既可用于预测石油馏分的物理化学性质,也可为下一步分子反应动力学模型的构建提供基础。但该方法的应用不是为了替代分析技术,而是弥补分析技术的不足。
4 结语与展望
当前,炼油行业正面临着产能过剩、国际环境变化多端、市场竞争白热化等严峻问题,同时还面临着炼油向化工转型的迫切需求,因此,高效合理地利用石油资源势在必行。只有充分认识石油原料的分子组成,掌握其转化成产品分子的规律,才能更好地提高竞争力。在现阶段还不具备对石油分子进行完整表征的前提下,以有限的分析数据,通过计算机模拟方法对石油分子进行重构正发挥着越来越重要的作用。尽管如此,石油分子重构技术依然需要进一步的优化和完善。
(1)构建更为准确的石油原料及产品分子库。一是构建更为合理的分子库:以当前最先进的分子水平表征平台为依托,通过分析大量的石油原料和产品,尽可能掌握石油原料及产品分子分布的统计学规律。二是选出更具代表性的分子库:对结构和性质相似的分子进行适当的归类和合并,将分子库的大小压缩至计算机容易处理的范围内。
(2)构建更为全面的石油原料及产品物性库。在充分考虑石油分子热力学性质、反应动力学性质的基础上,探究其分子结构与物理性质之间的关系,选取合适的混合规则和数学表达式计算石油原料及产品的宏观物性。
(3)选择合适的方法计算石油及产品的分子组成。依据分子库中分子数量的多少,选用适当的数学工具和优化算法,保证选取出来分子物性的定量计算结果的准确性。
以石油分子重构技术为依托,根据原料的分子组成及其结构特点,选择适宜的加工方案,使其转化成理想的目标分子,是今后石油加工发展的重要方向,也是实现石油分子高效转化的必由之路。
