RAG1基因新发变异致重症联合免疫缺陷1例报告及文献复习
2022-03-11崔清洋王梦斌曹银利唐成和
崔清洋,王梦斌,曹银利,唐成和
(新乡医学院第一附属医院 儿科,河南 卫辉 453100)
原发性免疫缺陷病(primary immunodeficiency diseases, PIDs)为一类和遗传紧密关联的异质性疾病, 患儿易患反复感染、肿瘤、过敏或自身免疫性疾病等。最新版的PIDs分类已在2020年1月17日由国际免疫学会联合会专家委员会发表,分为10大类,包括430种PIDs。其中同时影响细胞和体液免疫缺陷的第2亚类T-B-NK+SCID的原RAG1缺陷和RAG2 缺陷现在统称为RAG缺陷。
重症联合免疫缺陷病(severe combined immunodeficiency, SCID)是一类细胞免疫的缺陷、同时累及体液免疫的PID,SCID发生率约为1/100 000活产新生儿[1],而一项使用美国11个州的新生儿SCID筛查数据的研究发现,SCID发病率为1/58 000活产新生儿,包括典型SCID、泄漏型SCID和Omenn综合征[2]。此疾病一般具有明确的致病机制及干预治疗靶点,其遗传方式为常染色体隐性遗传,其中IL2RG基因变异所致 X 连锁联合免疫缺陷(X-linked severe combined immunodeficiency, SCID -XL)占 50%~60%(其中共用受体 γ 链基因缺陷是导致 SCID -XL最主要的形式,约占所有 SCID50%),重组激活基因(recombinase activating gene,RAG)变异(包括RAG1和RAG2)所致的联合免疫缺陷病占10%。SCID发病所涉及的基因有ADA、AK2、LIG4、NHEJ1、PRKDC、DCLRE1CRAG2、RAG1、CORO1A、LAT、CD247、CD3E、CD3D、PTPRC、IL7R、IL2RA、JAK3及IL2RG(其中IL2RG基因变异为X连锁,其余为常染色体隐性遗传)。
大部分SCID患儿出生时无明显异常,但随胎传抗体的消耗,患儿很容易在出生的第3~6月龄发生严重的反复感染,包括迁延性腹泻、口腔念珠菌感染、严重中耳炎、间质性肺炎及条件致病微生物如肺孢子虫和巨细胞病毒等感染,绝大部分患儿伴有生长发育迟缓[3]。若不及时纠正免疫缺陷,大部分患者在2岁以内因重症感染而死亡[4],为儿科医生必须面对的临床急症。
SCID通常基于缺陷的淋巴细胞亚群进行分类,可分为以下4类:T-B+NK+、T-B+NK-、T-B-NK+、T-B-NK-等,其中85%~90%的SCID患儿伴有已知的分子缺陷[5-6]。与T-B-NK+免疫表型有关的基因有DCLRE1C,LIG4,NHEJ1,PRKDC,XRCC4,RAG1,RAG2等,而T-B-NK+SCID约占SCID所有病例的25%。
本文回顾性分析1例婴儿SCID患儿的临床资料及基因检测结果,且RAG1基因的c.2428A>G杂合错义变异为国际首次报道及c.1229G>A杂合错义变异为国内首次报道,以期加强对本病的认识。
1 临床资料
女性患儿,1个月6天,因发现体质量不增半月余于2020年1月26日入院。半月余前发现患儿体质量不增,进乳可,大便4~5次/d,呈黄色糊状。G1P1,胎龄42周时因“胎儿宫内窘迫”剖宫产出生,出生体质量3 500 g,出生时有重度窒息史,羊水Ⅲ度污染,但无产伤史。出生1小时后因呼吸困难转入我院新生儿科,入院时血常规(2020-01-26)未见异常,见表1。予抗感染、营养脑细胞、静脉营养及合理喂养等治疗15天后好转出院。父母体健,非近亲结婚。入院体格检查:体质量3 500 g,一般情况尚可,发育不良,营养不良,体型偏瘦,心肺腹未见异常,四肢肌张力增高,四肢肌力正常。专科检查:语言及智能评估:会哭;仰卧位:头可居于中线,追视追听欠佳,双手不可居于中线位活动,双下肢屈曲,非对称型紧张性颈反射(ATNR)姿势存在;俯卧位:被动肘支撑,不可瞬间抬头;坐位:全前倾坐位;立位:踏步反射存在,双下肢可支撑体质量;手抓位:拇指内收;肌张力:四肢肌张力增高;关节活动度:股角60度,腘窝角120度,足背屈角30度,围巾征不过中线,跟耳征阴性。入院诊断:蛋白质-能量营养不良。辅助检查:甲状腺功能三项未见异常;血常规(2020-03-05)示粒细胞缺乏,见表1;大便常规潜血阳性;入院第2天患儿出现发热,超敏C反应蛋白(hs-CRP)22.31 mg/L;胸片示支气管肺炎;复查hs-CRP 43.05 mg/L,白细胞介素6(IL-6)48.67 pg/ml,降钙素原(PCT)0.90 ng/ml,连续复查2次血常规(2020-03-06和2020-03-09)均提示粒细胞缺乏或减少,见表1。予康复、先后予头孢替安及头孢他啶抗感染及降温对症治疗后患儿仍反复发热,升级抗生素为美罗培南加强抗感染后发热缓解,奶量可,但监测体质量有下降趋势,复查hs-CRP仍无明显下降,予丙种球蛋白(IVIG)增强机体免疫力,因不排除血液系统恶性疾病,进一步检查:自身抗体14项阴性,人微小病毒B19-DNA、EBV-DNA及人巨细胞病毒DNA均阴性。血培养及骨髓培养均阴性;网织红细胞绝对计数76.81×109/L,网织红细胞百分比0.03;骨髓穿刺结果示:①骨髓有核细胞增生活跃,粒系占66%,红系占22%,粒:红=3.0;②粒系增生活跃,比值增高,部分细胞胞体胀大,浆内颗粒增多增粗,中性分叶核粒细胞缺如;③红系增生活跃,比值及形态可;④淋巴细胞及单核细胞比例增高;⑤血小板易见。乳酸2.9 mmo/L;血氨34.8 mmo/L;血串联质谱及尿有机酸分析未见异常;复查肝功能:丙氨酸转氨酶(ALT)365 U/L,天冬氨酸转氨酶(AST)302 U/L,谷氨酰转肽酶150 U/L,总胆汁酸(TBA)17.16 μmol/L;淋巴细胞亚群示:淋巴细胞3.66%(40%~60%),总T淋巴细胞(CD3+)56.07%(60%~84%),CD3+CD8+19.53%(20%~35%),CD3+CD4+36.26%(36%~55%),CD3+CD4+CD8+0.31%(0~5%),CD3+CD4+/CD3+CD8+1.86%(0.8%~2.6%),CD19+B淋巴细胞41.48%(8%~28%),CD3+CD56+T-NK细胞1.96%,淋巴细胞计数430×106/L,总T淋巴细胞(CD3+)241×106/L,CD3+CD8+84×106/L,CD3+CD4+156×106/L,CD19+B淋巴细胞13×106/L ,CD3+CD56+T-NK细胞178×106/L,符合T-B-NK+免疫表型。排除白血病及再障和遗传代谢病,予调整抗生素、保肝及粒细胞刺激因子升高粒细胞治疗后患儿未再发热,体质量缓慢增加,白细胞升高(但容易再次下降),住院期间患儿出现腹胀,腹部X线立卧位平片示腹部部分肠管扩张并积气;予生理盐水回流灌肠腹胀减轻后钡剂灌肠提示直肠痉挛狭窄,建议隔期复查除外先天性巨结肠。住院38天后患儿家属要求出院转上级医院进一步治疗。出院诊断:①重度热能营养不良;②支气管肺炎;③肝损伤;④不完全性肠梗阻;⑤重度贫血;⑥脓毒血症;⑦中毒性肠麻痹;⑧先天性巨结肠?⑨直肠狭窄。
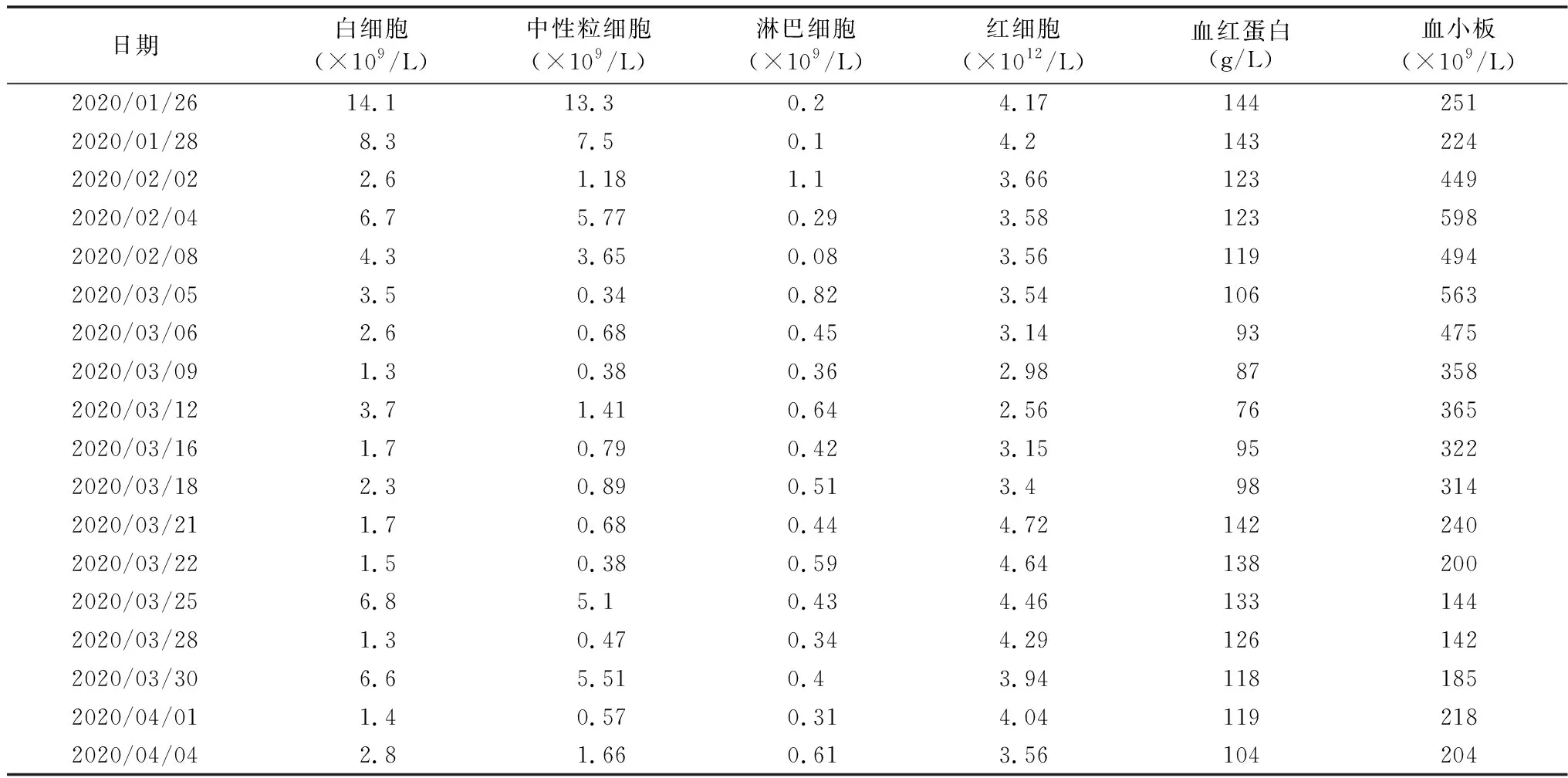
表1 血常规变化情况
因患儿多次血常规提示粒细胞减少甚至缺乏,考虑先天性中性粒细胞缺乏症可能,在获得家长知情同意后,采集患儿及父母的外周血行基因检测。RAG基因exon2引物为:正向引物序列 CCTGGAGAGTC CAG TG AAG TCC; TGCCAGGCTACC AC C ACTTT;反向引物序列 AGCCAG CAGG AACA AGGTCA; GAGAATGCCT CCC A GCTCAA。扩增条件为:①预变性:95 ℃,10 min;②变性(35个循环):95 ℃,30 s;③退火(35个循环):60 ℃,30 s;④延伸(35个循环):72 ℃,45 s;⑤彻底延伸(35个循环):72 ℃,5 min。
测序分析发现患儿RAG1基因第2外显子分别为存在c.1229G>A(p.Arg410Gln)、c.2428A>G(p.Ile810Val)错义变异(母源性)和c.2005G>A(p.Glu669Lys)的错义变异(父源性),见图1~3。上述变异可能导致蛋白质功能受到影响,c.1229G>A及c.2005G>A变异的致病性已有文献报道,但c.2428A>G变异的致病性未见报道(所参考数据库: HGMD Pro、 PubMed及ClinVar),且c.1229G>A杂合变异为国内首次报道。上述的变异不属于多态性变化,在人群中发生频率极低(所参考数据库 1000Genomes及dbSNP)。家系验证结果显示,患儿c.1229 G>A和c.2428A>G来自母亲,c.2005G>A来自父亲,为复合杂合变异,符合常染色体隐性遗传规律。

图1 RAG基因c.1229G>A变异测序峰图 a、b及c分别为患儿和父母,箭头示突变位点

图2 RAG基因c.2428A>G变异测序峰图 a、b及c分别为患儿和父母,箭头示突变位点
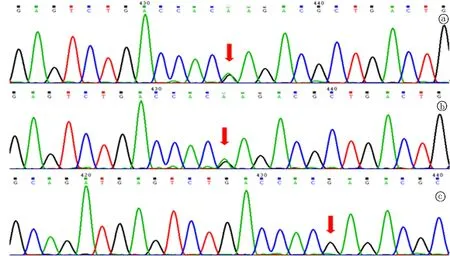
图3 RAG基因c.2005G>A变异测序峰图 a、b及c分别为患儿和父母,箭头示突变位点
根据文献[7]进行致病性分析:①RAG1基因 c.1229G>A变异的证据强度为“PS1+PM1+PM2+PM3+PP3”,判断为致病性的变异;②RAG1基因c.2005G>A变异的证据强度为“PS1+PM2+PM3+PP3+PP5”,判断为致病性的变异;③RAG1基因c.2428A>G变异的证据强度为“PM2+PM3+PP3”,判断为临床意义不明的变异,但亦未有无致病性的定论,需待日后进一步行相关验证。
RAG1基因序列全长为1 043个氨基酸,包括478个alpha-helix,121个extended strand,56个beta-turn,388个Random coil。各种二级结构所占的百分比为alpha-helix占45.83%,extended strand占11.6%, beta turn占5.37%,random coil占37.2%。c.1229G>A(p.Arg410Gln),导致第410号氨基酸由精氨酸变成了谷氨酰胺;c.2428A>G(p.Ile810Val),导致第810号氨基酸由异亮氨酸变成了缬氨酸;c.2005 G>A(p.Glu669Lys),导致第669号氨基酸由谷氨酸变成了赖氨酸。该3个变异均发生在RAG1蛋白家族结构域中,RAG1蛋白家族是RAG1-RAG2 V(D)J复合酶复合物的两个不同组分之一。RAG复合物由RAG1蛋白和RAG2蛋白组成,是一种多蛋白复合体,在V(D)J重组过程中介导DNA裂解,其中RAG1介导DNA-binding结合到保守重组信号序列(RSS)上,氨基酸的改变可能导致蛋白质功能受到影响,见图4~5。
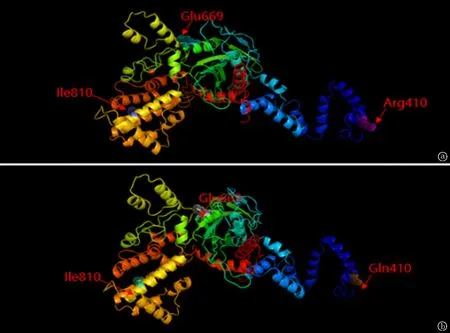
图4 c.1229G>A、c.2428A>G及c.2005 G>A变异位点(b)及变异前的位置(a)

图5 c.1229G>A、c.2428A>G及c.2005 G>A变异位点(b、d及f)及变异前的位置(a、c及e)
结合患儿临床表现及基因检测分析诊断RAG1基因c.2005G>A、c.1229G>A及c.2428A>G所致的SCID基本明确,告知患儿家属目前该病最有效的治疗措施为造血干细胞移植,家属考虑中,随访患儿一般情况暂时尚可。
2 讨 论
RAG1基因定位于11p12,基因组全长约11.73 kb,包含2个外显子,外显子长度约6 564 bp,编码1 043个氨基酸。目前RAG1基因变异类型多为错义变异,其次为无义变异、碱基缺失和插入变异,尚无变异类型与临床类型关系报道,但临床上纯合变异较复合杂合变异症状重,本例患儿RAG1基因3个变异位点均为错义变异,且予以升级抗生素及IVIG和粒细胞刺激因子治疗后症状改善,支持文献报道。RAG1基因氨基末端区域有核定位的信号,尤其是4个碱性区域BI、BIIa、BIIb及BIII,是核转运蛋白SRPI的靶点。另外2个锌指结构域C3HC4及C2H2参与RAG1蛋白二聚体的形成。RAG1蛋白的中心区域有2个显著保守的功能域,1个氨基酸残基区域,识别重组信号序列(RSSs),将RAG1-RAG2的蛋白复合体锚定在RSSs区。另一个氨基酸的残基区域是重组酶活性区域,与RAG2蛋白相互作用的功能域定位于其他氨基酸残基区域,包括活性中心和羧基末端区域。
RAG为Ig及TCR基因片段重排所必需,RAG1基因变异造成其编码的重组酶活性完全或部分丧失,V(D)J重组失衡,T及B淋巴细胞发育在早期为阻断而致PID发病。RAG1蛋白核心的氨基酸区域是387-1008,其编码产物Rag1蛋白所形成的复合物为最重要的重组酶之一。该基因完全变异造成功能丧失,阻断V(D)J重组,致成熟的B及T细胞功能完全丧失。
V(D)J基因片段的重组为RAG1或RAG2所诱发,其核酸内切酶诱导DNA双链断裂。由非同源的末端连接蛋白对断裂的DNA进行修复,该过程 至少包括5个蛋白,即Artemis、LIG4、XRCC4、DNA-PKcs和Ku70/80[8],这些分子的缺陷均可导致 T-B-NK+SCID发生。
如果不是新生儿筛查发现,T-B-NK+SCID患儿在生命的早期会出现严重到危及生命的感染、发育不良、T和B细胞数量和功能低下或缺失、NK细胞数量和功能正常。T-B-NK+SCID可由编码抗原受体基因V(D)J重组蛋白的任何一个基因的常染色体隐性缺陷引起。重组过程随机组合可变的、多样性的和连接的基因片段,这些片段分别编码TCR和免疫球蛋白基因。这些基因编码的一些蛋白质也参与了人体所有细胞的DNA修复。这些基因的缺陷与额外的免疫表型有关,包括生长发育异常以及对电离辐射和化疗的敏感性增高。
与V(D)J重组相关的T-B-NK+SCID主要有两种类型:有或无放疗/化疗敏感性的患者。其类型取决于缺陷基因是否同时参与V(D)J重组和DNA修复或重组。RAG1或RAG2基因的严重变异可致无辐射敏感性的T-B-NK+SCID,因RAG1和RAG2基因只参与V(D)J重组,而不参与DNA修复;某些RAG基因变异导致部分蛋白表达和T、B细胞产生数量受限但临床表现较为明显,称为Omenn综合征。Omenn综合征也为一种T-B-NK+SCID,尽管它是一种产生无功能T和B细胞的泄漏型SCID,因此可能表现为T+B+NK+SCID或轻度联合免疫缺陷。B-NK+SCID另一类型是辐射敏感性的SCID(RS-SCID),其中包括阿萨巴斯卡SCID(SCID-A),在讲阿萨巴斯卡语的美洲土著人中发现。RS-SCID主要由DCLRE1C基因变异引起,但也可以在涉及基因PRKDC、LIG4、NHEJ1和NBS1变异中发现,所有这些都是NHEJDNA修复所必需的。
SCID临床上可分为典型SCID,如果不太严重,则根据T细胞定性和定量缺陷的严重程度分为泄漏型SCID。新生儿筛查或家族史诊断为SCID的婴儿在出生时和婴儿早期表现正常。新生儿期未确诊的SCID患者的典型症状是反复严重感染、慢性腹泻和发育不良。在缺乏以人群为基础的新生儿SCID筛查的情况下,通常直到婴儿出现一种或多种严重感染才会做出诊断。大多数SCID患儿的胸片上没有胸腺阴影,淋巴细胞绝对计数通常很低。典型SCID定义为自体T细胞计数<300×106/L,体外淋巴细胞转化试验(PHA)反应<正常的10%,泄漏型SCID是指淋巴细胞减少(2岁以下的自体T细胞计数<1 000×106/L,2~4岁<800×106/L,或4岁以上<600×106/L),PHA反应<30%正常。该患儿自体T淋巴计数241×106/L,小于300×106/L,符合典型SCID定义,其在第1次住院时(即新生儿期)一般抗感染治疗有效,白细胞明显降低,考虑为一过性。第2次因发育不良住院,且在新生儿期之后出现不易控制的感染,支持上述文献报道。但该患儿胸片可见胸腺影,与上述文献略有不同。
对于SCID家庭及高危孕妇应加强遗传咨询,更重要的是进行必要的产前诊断,其一可早期采取措施减少SCID患儿的出生,其二也有利于医护人员为有缺陷患儿出生后提供及时而有效的诊治做好必要的准备。
SCID的初始管理为预防感染,包括与潜在的疾病接触者隔离、免疫球蛋白替代疗法、开始抗菌预防和避免使用活病毒疫苗。而大多数SCID最常见、最普遍及最有效的治疗方法是来自匹配良好的健康异基因供体的造血细胞移植。这种治疗有很好的整体生存率,重建T细胞免疫,在许多情况下,B细胞免疫及腺苷脱氨酶(ADA)缺乏可采用酶替代疗法。如果没有人类白细胞抗原(HLA)相同的供体,基因治疗在某些形式的SCID中是一种越来越有效和成功的替代方案。但遗憾的是,相当一部分患儿因经济和配型困难等原因,往往在造血细胞移植前死亡。
尽管依据美国医学遗传学与基因组学学会(ACMG)和美国分子病理学会(AMP)“基因序列变异的解释标准和指南”进行致病性分析显示尚无法明确c.2428A>G变异位点的致病性,但c.2428A>G不属于多态性变化,在人群中发生的频率极低(所参考的数据库 dbSNP及1000Genomes),为进一步明确RAG1基因c.2428A>G变异位点的致病性,需要进一步大样本回归分析并进行转基因动物模型验证。患儿有SCID典型的临床表现,故综合考虑基因结果及患者症状和家族史等信息,可确认c.2005G>A及c.1229G>A变异位点为患儿的致病性变异,扩充了SCID国际上的基因变异谱。
