甘肃甘南传统发酵牦牛乳中细菌菌群的多样性研究
2022-03-11马彩霞王湘竹
马彩霞 梁 琪,* 王湘竹 刘 瑛
(1甘肃农业大学食品科学与工程学院/甘肃省功能乳品工程实验室,甘肃 兰州 730070; 2甘肃临夏州燎原乳业有限公司,甘肃 临夏 731100)
甘肃甘南位于甘肃省西南部,地处甘、青、川三省交界地带,是甘肃藏区的重要代表,具有独特的地理环境和生物多样性[1-2]。甘肃甘南总面积约4.5万平方米,境内海拔1 100~4 900 m,紫外线强度高(单日紫外线指数可达到12.5),年平均气温约1.7℃,年降雨量约400~800 mm,属于典型的高原大陆季风气候[3-4]。畜牧业是甘肃甘南的支柱产业,现有草场4 000 余万亩,可利用面积约为3 800万亩[5]。牦牛属于牛亚科动物,对于高寒环境有着极强的耐受性,常生活在高海拔的青藏高原地区[6]。甘南牦牛生活在海拔2 800米以上的地区,主要以天然牧场牧草为食[7]。2015年统计数据显示,甘肃甘南牦牛总数约138万头,占全省牦牛总量的80%[5],为甘南牧民提供了丰富的牦牛乳资源。藏区牧民沿袭传统的发酵方法制作了多种牦牛乳制品,其中最常食用的是传统发酵牦牛乳[8]。甘肃甘南地区牧民制作的发酵牦牛乳历经数千年传承,在独特的自然环境下经过长期自然驯化使得其中蕴含丰富的微生物群落,因此将其作为研究对象极具代表性[9]。
近年来,高通量测序技术因具有成本低、测序质量高、能更客观全面地了解菌群结构等优势,可用于更好地评估微生物多样性,目前已广泛应用于发酵乳制品中微生物菌群结构及多样性的研究[10]。Shangpliang等[11]采用Illumina MiSeq高通量测序技术对印度阿鲁纳恰尔邦和锡金州的四大类共54份传统发酵乳制品中的细菌群落结构进行分析发现,不同种类的发酵乳制品细菌菌群多样性存在差异。Liu等[12]采用焦磷酸测序研究了俄罗斯卡尔梅克和赤塔州的蒙古族家庭的19种自然发酵牛乳(naturally fermented cow milk, NFCM)中细菌和真菌的群落多样性,结果表明卡尔梅克和赤塔州的样品存在菌群结构差异,地理环境的差异可能是影响两地样品中微生物多样性的重要因素。Sessou等[13]采用高通量测序技术对采自贝宁的手工自制奶酪样品,以及分别采自贝宁和尼日尔的20份发酵乳样品中酵母菌的多样性进行了研究,发现两国样品间的酵母组成存在显著差异。目前鲜有关于甘肃甘南地区传统发酵牦牛乳中细菌菌群多样性的研究报道,因此,全面了解该地区传统发酵牦牛乳中细菌菌群的多样性十分必要。本研究基于Illumina MiSeq高通量测序技术对采自甘肃甘南的传统发酵牦牛乳中细菌菌群的多样性进行表征,以期全面解析发酵牦牛乳中的细菌菌群结构,系统解析牦牛乳中的细菌组成及群落结构,为后期优良微生物资源的研究与开发奠定基础。
1 材料与方法
1.1 材料与试剂
样品于2019年7月采自甘肃省甘南藏族自治州合作市,勒秀乡麻拉行政村(海拔3 163.5 m)共采集4份(编号为M1、M2、M3、M4)、那吾乡加拉行政村(海拔2 889.4 m)共采集4份(编号为W1、W2、W3、W4)、那吾乡多河尔行政村(海拔3 000.5 m)共采集4份(编号为D1、D2、D3、D4)。样品QL1于2019年8月采自青海省海北藏族自治州祁连县(海拔3 015.4 m),所有样品均由牧民使用传统方法发酵制成。
TruSeqTMDNA试剂盒,美国Illumina公司;AP-MN-P-50凝胶提取试剂盒,美国Axygen Biosciences公司; MetaVxTM文库构建试剂盒,美国GENEWIZ公司。
1.2 仪器与设备
Qubit 3.0荧光计,德国Life公司;Illumina MiSeq测序仪,英国Illumina公司;7900HT型荧光定量 PCR仪,美国Applied Biosystems公司;Nanodrop 2000型超微量分光光度计,美国Thermo fisher公司;Agilent 2100生物分析仪,美国AgilenTTechnologies公司;5424R型高速台式冷冻离心机,德国Eppendorf公司。
1.3 试验方法
1.3.1 样品总DNA提取 使用DNA提取试剂盒依照说明书从13份样品中提取 DNA,用 Qubit®dsDNA HS Assay Kit 检测所提DNA浓度。
1.3.2 PCR扩增、文库构建及上机测序 以20~30 ng稀释后的样品总DNA为模板,以包含5′-CCT ACGG R R B GCA S CAG K V R V GAAT-3′序列的上游引物和包含5′-GGAC T AC N V GG GT W TC T AATCC-3′序列的下游引物对细菌16S rRNA的2个高度可变区(V3和 V4区)进行PCR扩增[14]。扩增体系(25 μL)为:TransStart Buffer 2.5 μL,dNTPs 2 μL,上下游引物各1 μL,模板 DNA 20 ng,用ddH2O补齐至25 μL。PCR 反应程序:95℃预变性2 min;95℃、55℃、72℃分别持续30 s,共30次循环;72℃延伸5 min,得到的产物用1.5%琼脂糖凝胶电泳检测完整性。PCR扩增完成后使用 MetaVxTM文库构建试剂盒构建测序所需文库。使用 Agilent 2100 生物分析仪检测构建完成的文库质量,再通过Qubit3.0荧光计检测文库的浓度[15]。将构建好的文库定量到10 nmol·L-1,按照Illumina MiSeq测序仪使用说明书进行PE250/FE300双端测序[16]。PCR 扩增、文库构建及测序由苏州金唯智生物科技有限公司完成。
1.3.3 测序相关分析 将双端测序所得的正反向reads进行两两组装,过滤去除组装结果中含有N的序列且保留序列长度大于200 bp的序列。所得的序列再经过质量过滤,去除嵌合体序列。使用VSEARCH(1.9.6)进行序列聚类,将97%的序列相似水平作为划分阈值,聚类成可操作分类单位(operational taxonomic units,OTU)[17]。基于OTU的分析结果,分别计算得到ACE指数、Chao1 指数、Shannon指数等α多样性指数,并绘制稀释曲线[18]。使用QIIME软件对基于非加权距离矩阵进行算术平均组对法(unweighted pair group method with artithe metic mean, UPGMA)聚类分析,用主坐标分析(principal co-ordinates analysis,PCoA)可视化图展示出样品的β多样性[19]。最后采用PICRUSt(V1.0)软件对细菌群落的基因功能特征进行预测分析[20]。
1.4 数据分析
利用Vsearch(1.9.6)、Qiime(1.9.1)、Metastats、 LEfSE(1.0)、Cutadapt(v1.9.1)、PICRUST(v1.0.0)等软件进行数据分析。
2 结果与分析
2.1 PCR扩增基本情况
由图1可知,13份样品的PCR 扩增产物凝胶电泳在500 bp左右出现了清晰明显的条带,说明目的片段扩增成功。
2.2 样品序列数特点
通过细菌的16S rRNA基因V3-V4 区测序,13份样品共产生720 221条高质量序列,平均长度为463 bp。高质量序列占有效序列的比例为94%。将所有的有效序列按照97%的相似度进行OTU 聚类,共聚成166个OTU。
注:1:M1;2: M2;3:M3;4:M4;5:W1;6:W2;7:W3;8:W4;9:D1;10:D2;11:D3;12:D4;13:QL1.图1 PCR扩增产物凝胶电泳图Fig.1 Electrophoresis of PCR amplified product
2.3 稀释曲线
稀释曲线表示随着测序量的增加,可能会检测到的物种种类随之增加的状况,被广泛用于判断测序是否达到一定的深度[21]。由图2可知,随着测序量的增加,样品M1、M2、M3、M4、W1、W2、W3、W4、D1、D2、D3、D4、QL1的OTUs逐渐增加后趋于平稳,表明测序深度充分,基本覆盖了样品中所有的物种。
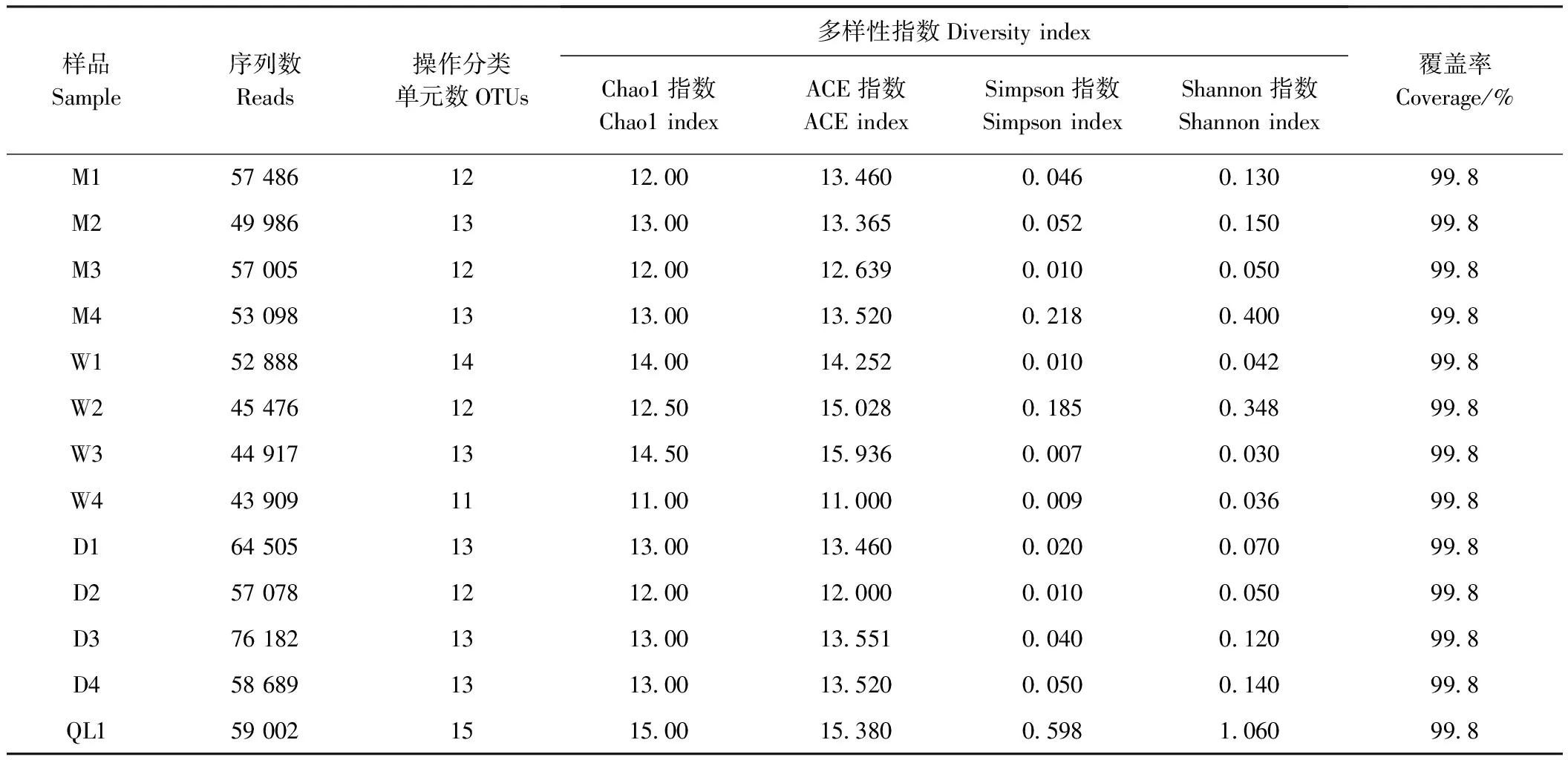
表1 各样品Alpha多样性指数Table 1 Alpha diversity index of each sample
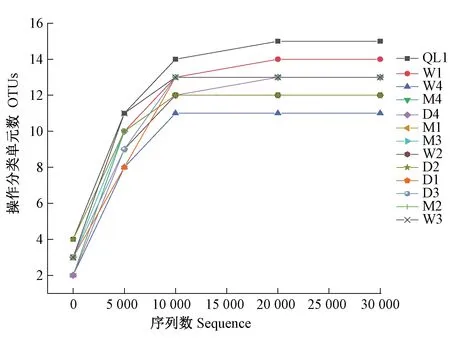
图2 各样品稀释曲线Fig.2 Rarefaction curves of each sample
2.4 Alpha多样性指数分析
Alpha多样性用一系列的统计学指数来评估样本的物种丰富度及多样性,主要包括Chao1指数、ACE指数、Simpson指数、Shannon指数等[22]。Shannon指数越高该样品的多样性越丰富。由表1可知,13份样品的多样性指数均不相同。样品Shannon指数从高到低依次为QL1>M4>W2>M2>D4>M1>D3>D1>M3=D2>W1>W4>W3,说明不同来源样品的细菌菌群多样性不同。
2.5 样品细菌菌群在门水平的分类
由表2可知,13份样品中共鉴定出4个细菌门,分别是厚壁菌门(Firmicutes)、变形菌门(Proteobacteria)、蓝菌门(Cyanobacteria)和拟杆菌门(Bacteroidetes)。厚壁菌门(Firmicutes)为13份样品中的优势门,13份样品的相对丰度介于99.95%~99.99%。变形菌门(Proteobacteria)为亚优势门,蓝菌门(Cyanobacteria)和拟杆菌门(Bacteroidetes)为低丰度门,这两类门在样品QL1中均未鉴定到。由门水平分类来看,不同样品在门水平由不同的菌落组成,对各菌门表现出不同的丰度。
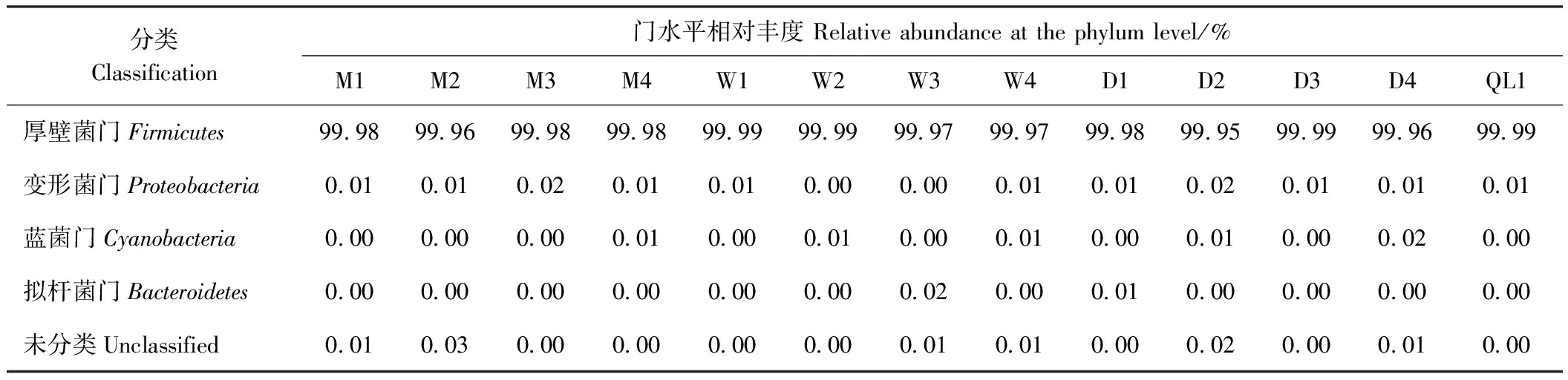
表2 各样品门水平菌群分布相对丰度Table 2 The relative abundance of horizontal flora distribution aTThe phylum level of each sample
2.6 样品细菌菌群在属水平的分类
由表3可知,13份样品共鉴定出了4种不同的细菌菌属以及在属水平上未鉴定的属(Unclassified)。其中,乳杆菌属(Lactobacillus)为高丰度菌属,链球菌属(Streptococcus)为第二丰度菌属,在13份样品中均检测到。鲁杰氏菌属(Ruegeria)和普雷沃菌属(Prevotella)为低丰度菌属,仅在部分样品(M4、D1、D4)中检测到,而在QL1中两种属均未检测到。表明不同样品在属水平的分类不同,对各属表现出不同的丰度值。
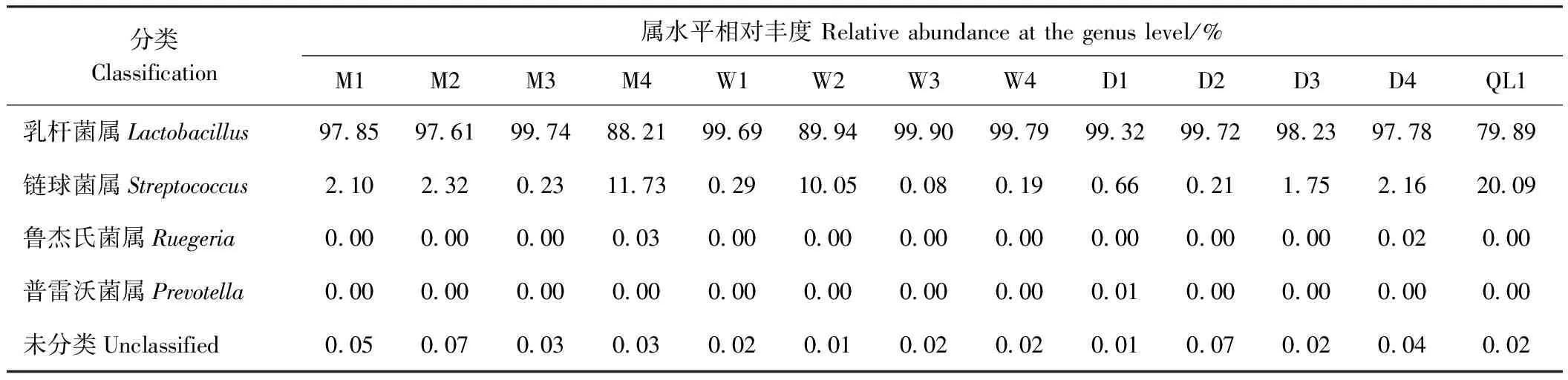
表3 各样品属水平菌群分布相对丰度Table 3 The relative abundance of horizontal flora distribution aTThe genus level of each sample
2.7 β多样性分析
β多样性是对不同样品中微生物群落组成的比较分析,从而反映出不同样本间的多样性的差异[23]。
2.7.1 主坐标分析 主坐标分析(PCoA)是通过R语言来分析样品间的相似性或者差异性[24]。13份样品的PCoA分析如图3所示,PC1-PC2为第一和第二主坐标得到的PCoA图,PC1-PC3为第一和第三主坐标得到的PCoA图。PC1、PC2、PC3对样品差异的贡献率分别为99.4%、0.38%、0.13%。样本之间距离代表了样本微生物群落的相似性,距离越近,相似度越高[25]。由图3可知,在PC1和PC2维度上部分样品交叠在一起,而在PC3维度上所有样品可以很好地区分。由样品PCoA分析可知,在PC2维度上W1、W3、W4、D1、D2、M3距离较近有着相似的群落, M1、M2、D3、D4距离较近有着相似的群落,W2和M4距离较近有着相似的群落,整体甘南地区的样品群落相似性较高,与QL1相距较远存在显著差异。
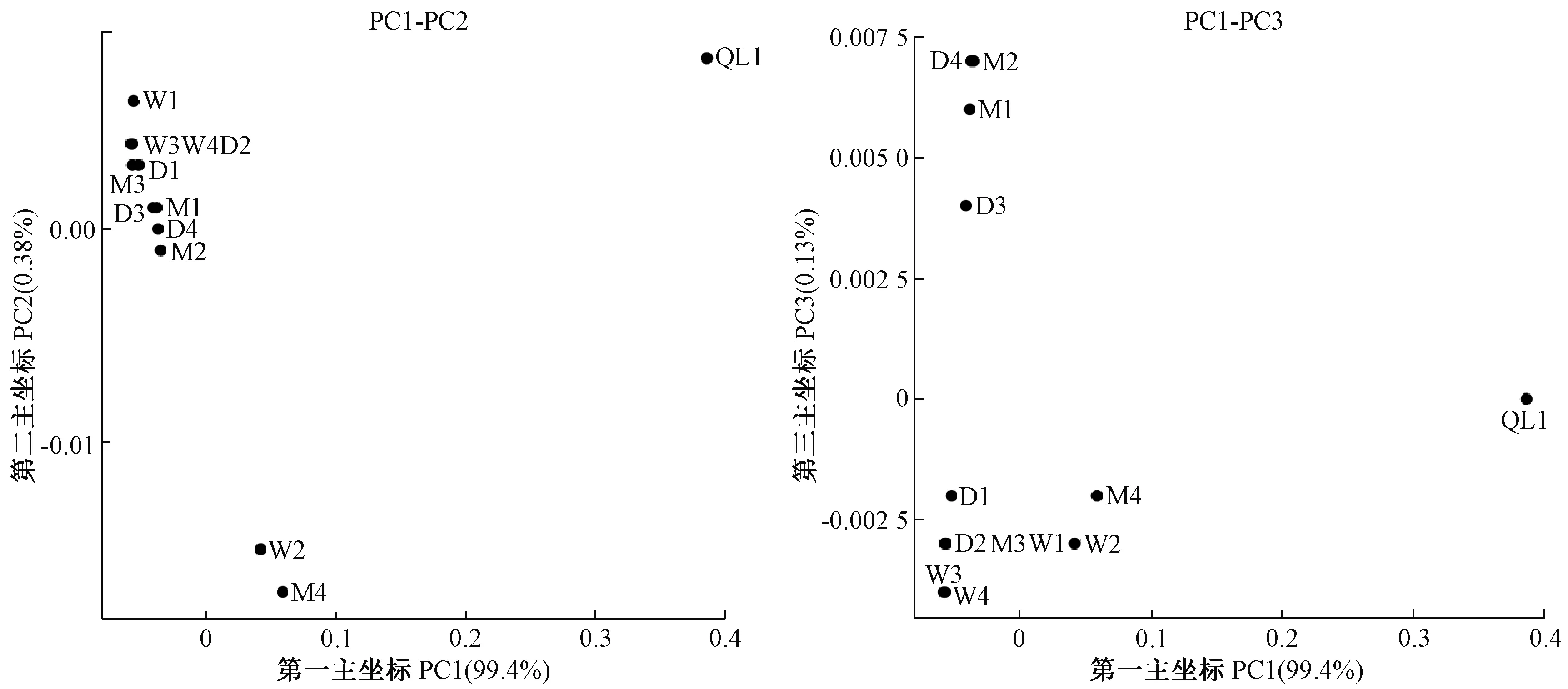
图3 各样品PCoA分析Fig.3 Principal co-ordinates analysis of each sample
2.7.2 样品菌群结构聚类分析 样品聚类分析是指使用算术平均组对法(UPGMA)分析样本在特定进化谱系中是否有显著微生物群落差异[26]。两样本越相似,样本间的分枝长度越短。由图4可知,13份牦牛酸乳样品聚为两大类,第一大类中W1、W3、W4、D1、D2、M3分支较小,相似度较高;M1、M2、D3、D4分支较小,相似度较高,W2和M4分支较小,相似性较高聚为一类。QL1单独聚为一类,表现出与甘肃甘南地区样品相比不同的微生物群落特征。表明甘南地区12份样品(W1、W3、W4、D1、D2、M1、M2、D3、D4)的菌群差异不显著,有着相似的进化谱系,而QL1单独聚为一类,与甘南地区样品差异较大,这与PCoA分析结果一致。
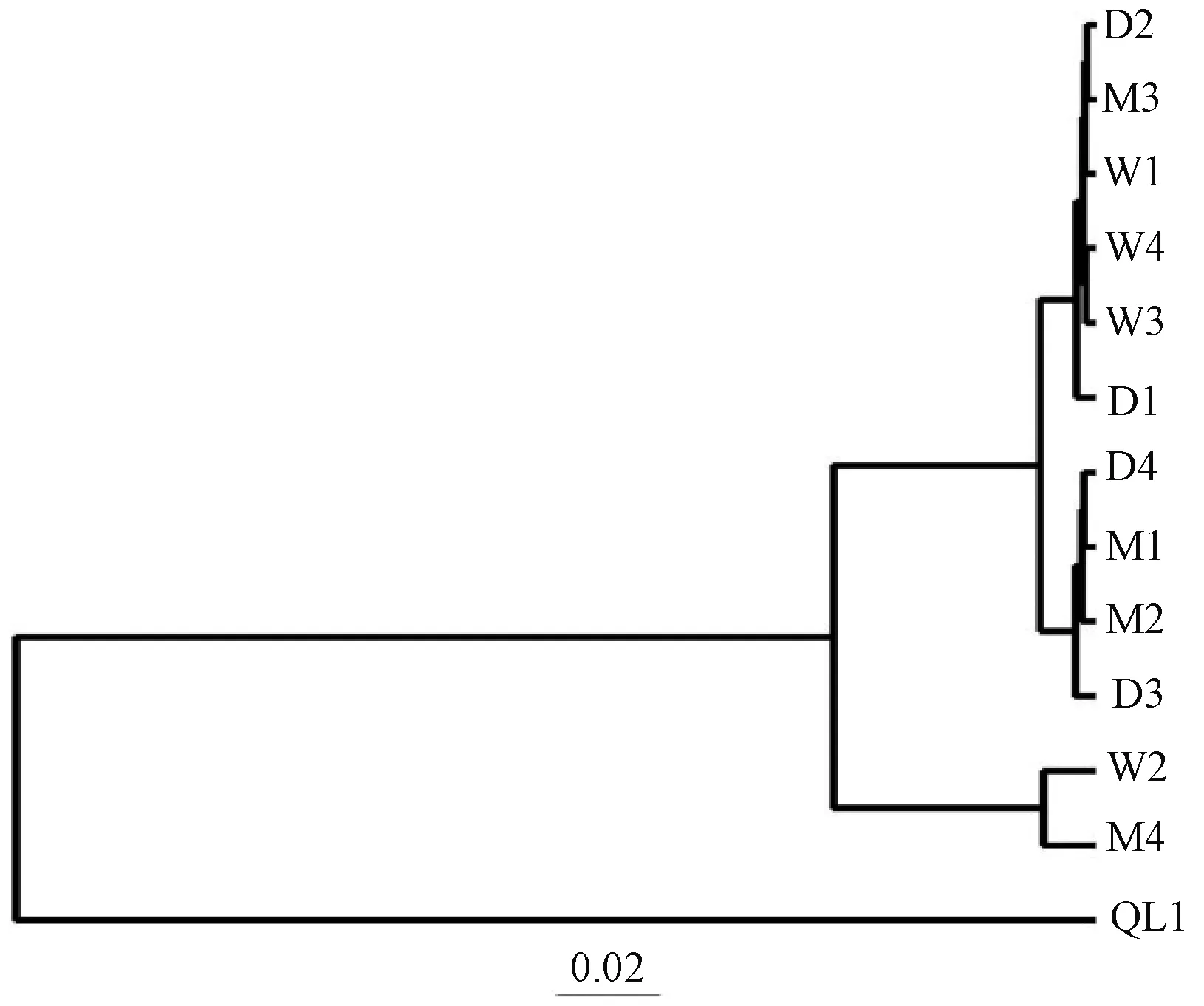
图4 各样品UPGMA菌群结构聚类分析Fig.4 UPGMACluster analysis of the bacterial communities in each sample
2.8 样品的PICRUSt功能预测分析
PICRUSt软件(phylogenetic investigation of communities by reconstruction of unobserved states),将测序得到的菌群组成“映射”录入Greengenes数据库中,实现对菌群代谢功能的预测。目前已经广泛用于微生物生态学[27]。由表4可知,在13份样品的功能基因中,基本预测功能占主导地位,占所有基因的11.76%,其次占比较多的是复制/重组/修复、碳水化合物代谢相关的基因,分别占所有基因的9.54%、9.39%。
3 讨论
本研究基于Illumina MiSeq高通量技术对甘肃甘南传统发酵牦牛乳中细菌菌群的多样性进行分析,结果表明,不同来源的传统发酵牦牛乳样品中细菌菌群的结构存在一定差异。本研究使用的发酵牦牛乳是通过传统方法生产的,因此样品间细菌群落结构的差异可能与原料、地理位置和其他环境因素有关[28-29]。在门水平上,厚壁菌门(Firmicutes)是优势菌门,每个样品表现出不同的丰度,此外还鉴定出低丰度的拟杆菌门和蓝菌门,这与新疆塔城地区发酵骆驼乳[30]、新疆昭苏县和特克斯县发酵酸牛奶[31]、云南地区发酵山羊奶制品[32]的研究结果一致。在属水平上,乳杆菌属(Lactobacillus)为高丰度菌属, 链球菌属(Streptococcus)为第二丰度属,这两个属通常与天然发酵乳制品有关,在乳制品的生产发酵过程中发挥着重要作用[33-34]。据报道,乳杆菌属的酸耐受能力高于链球菌属和乳球菌属[35]。在牦牛乳发酵过程中,随着酸度的上升,高比例的乳酸杆菌被保留富集,因此乳杆菌属成为高丰度的菌属。此外,在部分样品中还出现了低丰度的鲁杰氏菌属(Ruegeria)和普雷沃菌属(Prevotella)。鲁杰氏菌属最早由Uchino等[36]于1988年引入,属于有益细菌属,具有较高的磷酸二酯酶和磷酸单酯酶活性,在水生生物体体内大量存在,但在以往的发酵乳制品中未见该属的相关报道[37]。普雷沃菌属来自拟杆菌门,在厌氧环境中繁殖,是肠道菌群的主要组成部分,有助于降解食物中的碳水化合物和多糖,参与合成多种维生素等[38]。Quigley等[38]利用高通量测序技术揭示了62种爱尔兰手工奶酪中的细菌群落,结果发现,奶酪在发酵过程中有几个菌属参与,其中就包括普雷沃菌属,分析原因是原料乳中带入了普雷沃菌属。因此推断本研究中甘南地区样品检测出的低丰度的鲁杰氏菌属和普雷沃菌属可能来源于原料乳或发酵环境。
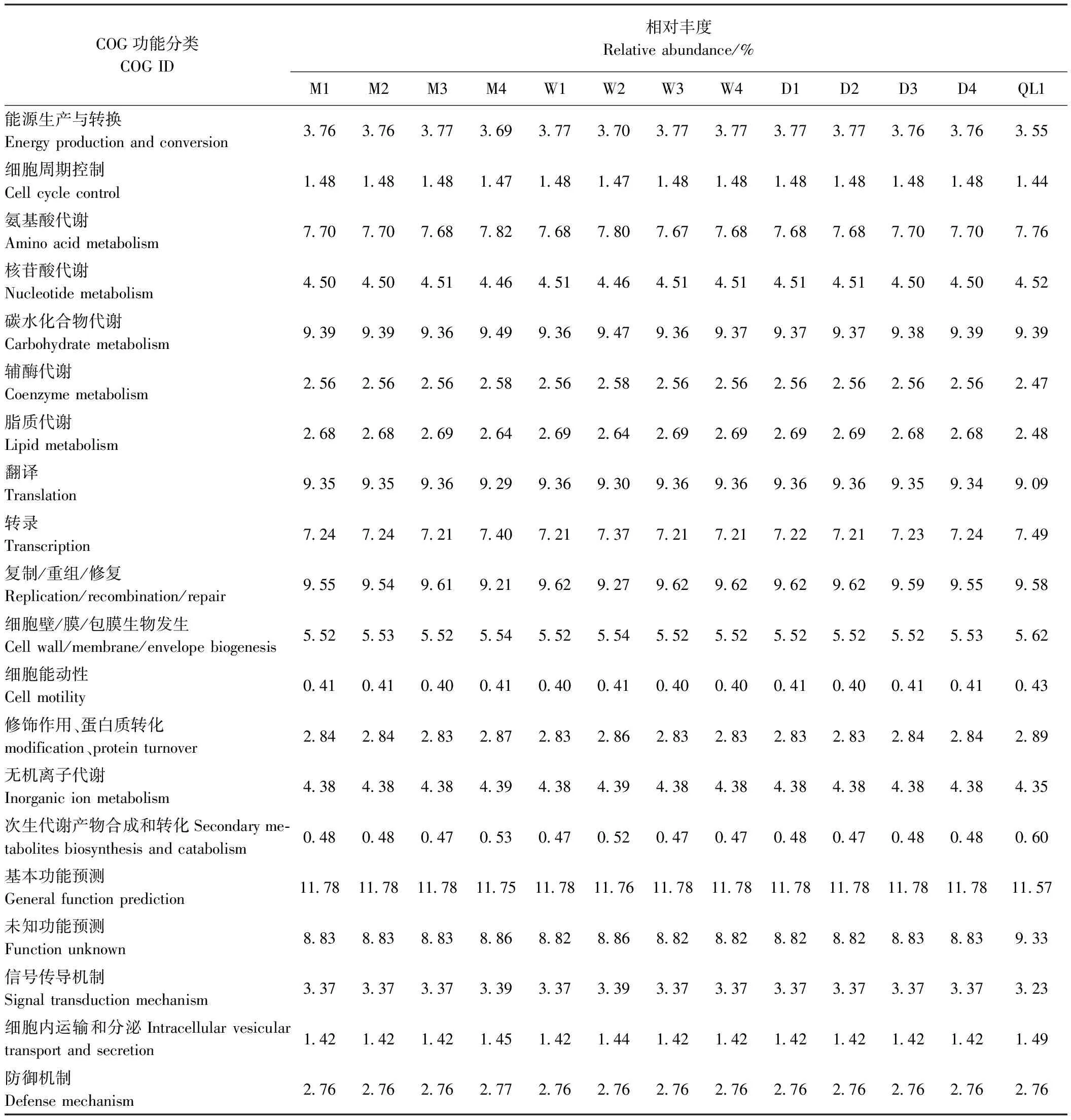
表4 各样品COG功能分类相对丰度统计表Table 4 Statistical table of relative abundance of COG functional classification of each sample
有学者基于PICRUSt对哺乳动物肠道微生物菌群的功能进行综合评估,发现哺乳动物体内的部分代谢与肠道微生物有关[39-40]。在本研究中,基于每个样品的16S rRNA测序数据进行PICRUSt分析发现,基本预测功能占主导地位,占所有基因功能的11.76%。其次占比较多的是复制/重组/修复、碳水化合物代谢相关基因,分别占所有基因的9.54%、9.39%。正是存在于乳酸菌中的各个功能基因,乳酸菌表现出了优良的特性。
4 结论
本研究基于Illumina MiSeq高通量技术对甘肃甘南地区传统发酵牦牛乳进行细菌菌群的多样性分析。结果表明,甘肃甘南地区传统发酵牦牛乳中具有丰富的细菌多样性,优势菌门为厚壁菌门(Firmicutes),优势菌属为乳杆菌属(Lactobacillus),不同来源的发酵牦牛乳在门属水平呈现不同的丰度。β多样性分析结果表明其群落相似性较高,有相似的进化谱系。通过预测功能基因,发现传统发酵牦牛乳中的绝大部分细菌与基本预测功能有关。
