动物源性食品中16种β-受体激动剂高灵敏检测与膳食暴露评估
2022-03-09牛秀丽王君孙世琨苏阿龙刘煜
牛秀丽 王君 孙世琨 苏阿龙 刘煜

摘 要:为有效开展食品安全风险评估,对β-受体激动剂(瘦肉精)进行有效监管,本研究以超高效液相色谱-串联质谱(UPLC-MS/MS)联用法建立了动物源性食品中16种痕量β-受体激动剂的检测方法。经酶解、提取、固相萃取净化,采用多反应监测(MRM)同时捕集16种目标分子母离子-子离子离子对,7.5 min内实现高效、准确分离测定。16种目标物加标回收率为64.5%~112.3%,日间精密度0.62%~1.37%,保留时间偏差为0.010~0.042,检出限为0.05~0.84 ?g/kg,定量限為0.17~2.76 ?g/kg。应用本方法完成261批动物源性食品的安全风险监测,实现快速准确的批量筛选,发现21批样品中残留11种β-受体激动剂。依据风险监测研究结果对β-受体激动剂进行膳食暴露评估,发现当同一动物组织中同时含有多种受体激动剂,总含量超过1.08 ?g/kg时将增加健康风险。
关键词:β-受体激动剂;痕量检测;膳食暴露评估;莱克多巴胺
Abstract: In order to effectively carry out food safety risk assessment and effectively supervise for the β-agonists, a method for the detection of 16 trace β-agonists in animal-derived food was established by ultra-high performance liquid chromatography- tandem mass spectrometry (UPLC-MS/MS). After enzymatic hydrolysis, extraction, and solid-phase extraction purification, 16 target molecular precursor ion-product ion ion pairs were simultaneously captured by multiple reaction monitoring (MRM), and accurate separation and determination were achieved within 7.5 minutes. The recovery rates of 16 target compounds were 64.5%~112.3%, the inter-day precision was 0.62%~1.37%, the retention time deviation was 0.010~0.042, the detection limit was 0.05-0.84 ?g/kg, and the quantification limit was 0.17~2.76 ?g/kg. Using this method, the safety risk monitoring of 261 batches of animal-derived food was completed. Fast and accurate batch screening was achieved, and 11 β-receptor agonists were found in 21 batches of samples. Dietary exposure to beta-agonists was assessed based on the results of risk surveillance studies. Increased health risk was found when multiple receptor agonists were present in the same animal tissue at a total level of more than 1.08 ?g/kg.
Keywords: β-receptor agonist; trace detection; dietary exposure assessment; ractopamine
“瘦肉精”是指肾上腺素受体激动剂类(β-兴奋剂),主要包括盐酸克仑特罗、莱克多巴胺、沙丁胺醇等。β-兴奋剂类既不是兽药,也不是饲料添加剂,其可有效促进动物的肌肉生长,提高动物酮体的瘦肉率并使其增重。β-兴奋剂易蓄积在动物体内,食用含有残留肾上腺素受体激动剂的动物源性食品会造成人体的急性或慢性中毒,严重者会有生命危险,威胁生命安全。从对食品安全的管控和消费者健康角度出发,国内禁止使用“瘦肉精”类药物用于食品动物的饲养生产过程,但在动物饲养过程中“瘦肉精”仍屡禁不止[1-4],2021年3月15日,央视“3·15”晚会再次曝光了“瘦肉精羊”事件。
对于β-受体激动剂的分析技术,国内外相关研究通常采用液相色谱-质谱联用法[5-6]、气相色谱-质谱联用法[7]、毛细管电色谱耦合质谱法[8]、高效液相色谱法[9]、免疫分析法[10]、毛细管电泳法[11]、电化学传感器方法[12-13]和表面增强拉曼光谱法[14]等。由于液相色谱-串联质谱法准确度高、无假阳性、检出限低、重现性好和无需衍生等特点,β-受体激动剂类药物残留检测国家标准主要采用液相色谱-质谱联用法。然而,由于肾上腺素受体激动剂品种多、化学结构和性质各异、待测组分复杂,残留水平一般较低和烦琐冗长的样品前处理过程一直干扰着检测结果的准确性,因此需要更高效灵敏的前处理技术和检测技术实现对其准确快速的测定。本研究利用固相萃取处理与超高效液相色谱-串联质谱技术,实现对16种β-受体激动剂的准确、快速、定量检测分析,并开展受体激动剂膳食暴露研究。
1 材料和方法
1.1 仪器与试剂
仪器:超高效液相色谱-串联质谱联用仪(Q TRAP 5500 AB Sciex Instruments);均质器(SilentCrusher M,德国海道夫公司);涡旋混合器(VORTEX 3,德国IKA);超速离心机(Sorvall LYNX6000,Thermo Scientific);氮吹仪(N-EVAP-45,Organomation);恒温摇床(MaxQ 420 HP,Thermo Scientific);正压固相萃取仪(SPE,J2 Scientific);超声波清洗器(Elmasonic P120H,Elma);超纯水系统(Thermo Scientific);-40 ℃冰箱(HYCD-282,青岛海尔);电子天平(XSE 105DU,梅特勒托利多)。
标准样品:沙丁胺醇(99.9%)、特布他林(99.0%)、盐酸克伦特罗(99.0%)、盐酸莱克多巴胺(96.1%)、西马特罗(99.9%)、盐酸多巴胺(99.0%)、马布特罗盐酸盐(>98.0%)、氯丙那林(99.6%)、非诺特罗(99.0%)、马喷特罗盐酸盐(>98.0%)和酒石酸福莫特罗(98.2%),上述11种标准样品均购自Dr.Ehrenstorfer GmbH公司;盐酸溴布特罗(99.0%)和苯乙醇胺A(99.0%)购自天津阿尔塔科技有限公司,盐酸妥布特罗(99.0%)和苯氧丙酚胺盐酸盐(98.0%)购自Bepure公司,盐酸齐帕特罗(98%)购自南京生利德生物科技有限公司。
试剂:β-葡萄糖醛甙酶/芳基硫酸酯酶(10 000 units/mg,MERCK);甲酸、甲醇、乙腈(MERCK色谱纯试剂),其他试剂均为分析纯;固相萃取柱:MCX强阳离子交换SPE柱(60 mg,3 mL,Waters)。
1.2 试验方法
1.2.1 β-受体激动剂标准溶液配制
准确称取适量沙丁胺醇、特布他林、盐酸克伦特罗、盐酸莱克多巴胺、西马特罗、盐酸多巴胺、马布特罗盐酸盐、氯丙那林、非诺特罗、马喷特罗盐酸盐、酒石酸福莫特罗、盐酸溴布特罗、苯乙醇胺A、盐酸妥布特罗、苯氧丙酚胺盐酸盐和盐酸齐帕特罗标准品,用甲醇溶解并分别配制成100 ?g/mL的单一物质β-受体激动剂储备液,避光保存于-18 ℃冰箱内,有效期12个月。准确移取0.5 mL上述16种单一物质标准储备液至50 mL容量瓶中,用甲醇稀释定容,配制成1.0 ?g/mL 16种β-受体激动剂混合标准储备液。工作液现用现配,使用前用流动相(0.1%甲酸-乙腈,96∶4,V/V)由混标储备液逐级稀释,配制成浓度范围为1.42~276 ?g/kg的混合標准工作液。
1.2.2 仪器分析
(1)色谱条件。Waters ACQUITY UPLC BEH C18色谱柱(100 mm×2.1 mm,1.7 ?m);柱温:40 ℃;样品室温度:4 ℃;进样体积:5.0 ?L;流动相A:0.1%(V/V)甲酸水溶液;流动相B:乙腈;流速:0.30 mL/min;梯度洗脱条件:0.00~2.00 min,4% B;2.00~10.0 min,4%~65% B;10.00~10.11 min,65%~4% B。
(2)质谱条件。离子化方式:电喷雾(ESI)离子源;正离子多反应监测(MRM)扫描;气帘气:35 psi;碰撞气:Medium;喷雾电压:5 500 V;雾化温度:550 ℃;雾化气:55 psi;辅助加热气:50 psi。试验所用脱溶剂气为高纯氮气,碰撞气为高纯氩气,使用前调节各气体流量以使质谱灵敏度达到检测要求。
1.2.3 样品制备
称取2 g(精确到0.01 g)样品于50 mL离心管中,加入8 mL乙酸钠溶液,再加50 ?L β-葡萄糖醛甙酶/芳基硫酸酯酶,充分混匀,37 ℃水浴水解12 h。水解完成后高速冷冻离心10 min,转数22 000 r/min,冷冻离心温度-10 ℃。离心完毕,取出离心管去除油脂层,将上清液全部转移至洁净离心管中,再次高速冷冻离心去除油脂层。最后,准确移取4 mL上清液转移至50 mL离心管中,加入10 mL饱和氯化钠溶液和10 mL异丙醇-乙酸乙酯(6+4)混合溶液充分提取;提取完成后7 000 r/min离心10 min,转移全部有机相,40 ℃水浴下氮气吹干,加入5 mL乙酸钠缓冲液,超声至充分溶解,待净化。
MCX固相萃取柱先用3 mL甲醇和3 mL水活化,将上述残渣溶液全部过柱,然后依次用3 mL水、3 mL 2%甲酸水溶液和3 mL甲醇淋洗,氮气加压抽干。用5 mL 5%氨水-甲醇溶液洗脱。收集洗脱液,在40 ℃水浴下氮气吹干,残余物用流动相1.0 mL溶解后,涡旋混匀,过0.22 ?m微孔滤膜,供UPLC-MS/MS测定。
2 结果与分析
2.1 16种β-受体激动剂检测质谱参数的确定
利用针泵进样将16种β-受体激动剂的混合标准溶液注入质谱仪中优化质谱参数。目标物进入电喷雾离子源后,均能够形成稳定的加合离子,以ESI正离子模式对激动剂药物进行母离子确认及子离子全扫描。本方法根据待测目标物的保留时间,采用分段扫描的方式对不同药物的特征离子进行数据采集,以提高灵敏度。16种β-受体激动剂的保留时间及定性、定量离子、去簇电压和碰撞能量等质谱条件见表1。
2.2 方法学验证
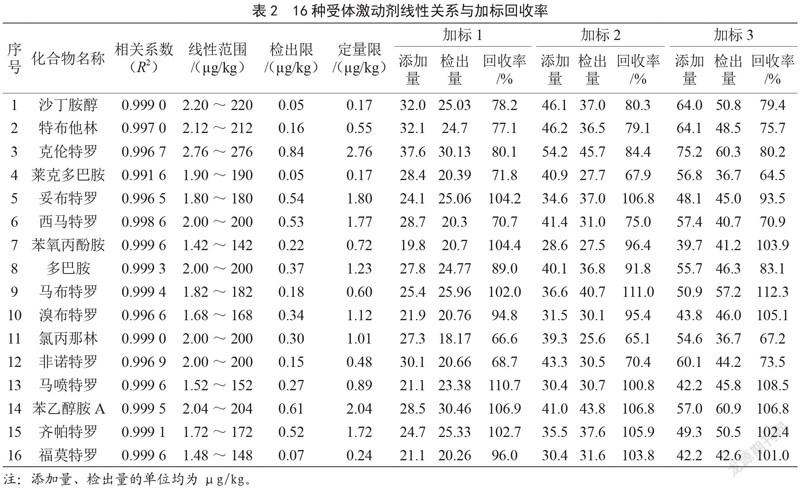
移取1.0 ?g/mL的β-受体激动剂混合标准储备液,用流动相(乙腈-0.1%甲酸水溶液,4∶96,V/V)配制成系列浓度的混合标准溶液,以各组分的浓度与其定量离子峰面积进行线性回归。由表2可知,各受体激动在1.42~276 ?g/kg浓度范围内呈良好的线性关系(R>0.991),检出限为0.05~0.84 ?g/kg、定量限为0.17~2.76 ?g/kg。
回收率及精密度试验以空白猪肉为待测样品,准确称取3份猪肉空白样品,添加低、中、高3个不同水平的16种β-受体激动剂混合标准溶液,进行前处理,测定目标化合物。16种β-受体激动剂在3个添加水平下的回收率为64.5%~112.3%。同一浓度样品日间精密度相对标准偏差为0.62%~1.37%,保留时间偏差为0.010~0.042。本方法从选择性、灵敏性、稳定性等方面均适用于动物源性食品中16种β-受体激动剂高灵敏检测,满足国内外法规对β-受体激动剂残留检测要求的执行限量。
2.3 实际样品检测
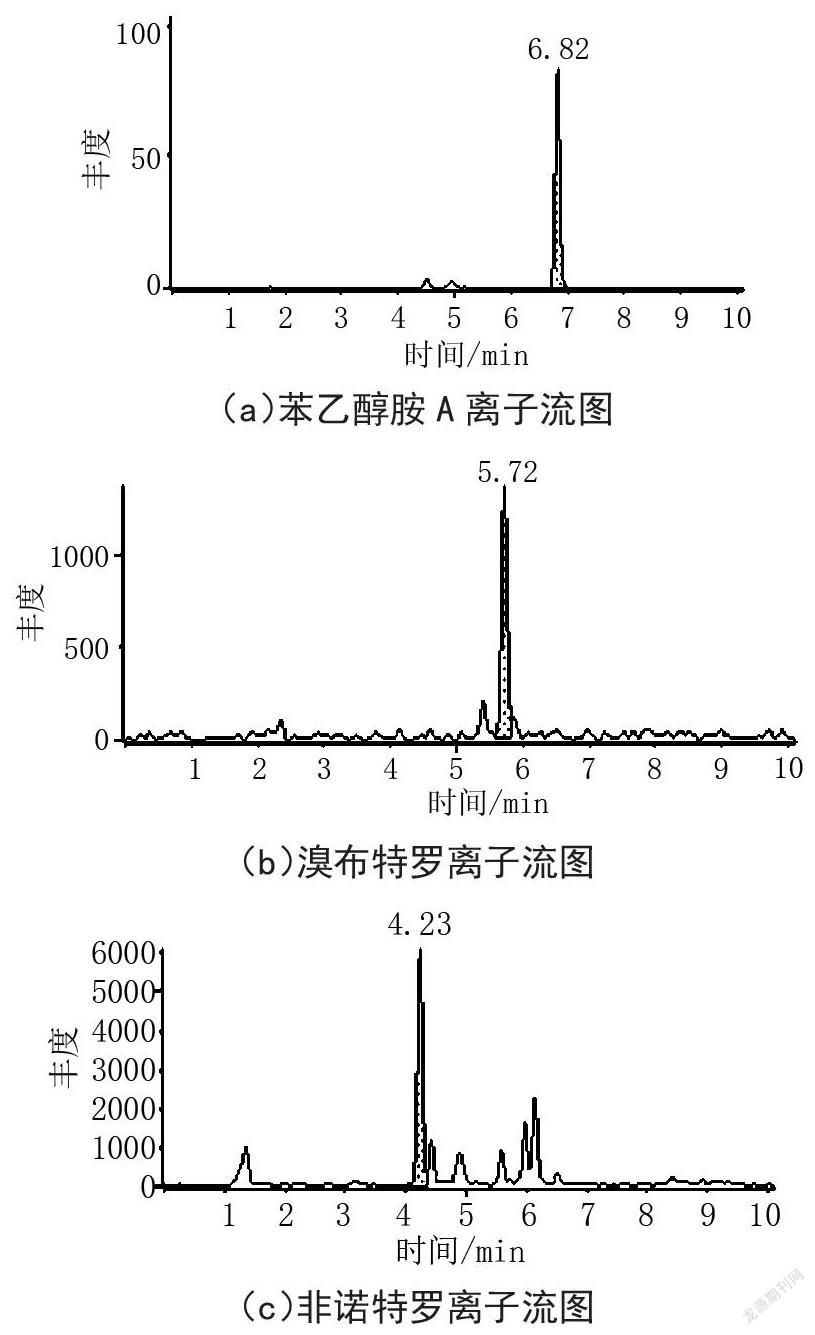
采集动物源性食品样品猪肉160批,猪肝脏31批,牛肉27批,羊肉26批,羊肾脏5批,猪心脏12批,总计261批。按照本文前处理方法与超高效液相色谱-串联质谱联用分析技术对采集样品进行测定,检测到21批样品中有β-受体激动剂的存在,总体检出率约为8.05%,其他240批样品未检出β-受体激动剂。其中检测到11种β-受体激动剂的残留及含量分别为:莱克多巴胺0.17~4.48 ?g/kg、多巴胺2.98~3.04 ?g/kg、马布特罗2.99 ?g/kg、非诺特罗1.78 ?g/kg、苯乙醇胺A 36.23 ?g/kg、齐帕特罗3.40~6.08 ?g/kg、溴布特罗4.74~5.37 ?g/kg、福莫特罗0.54 ?g/kg、氯丙那林3.34 ?g/kg,沙丁胺醇和特布他林有检出但低于定量限0.17 ?g/kg和0.55 ?g/kg。结果表明动物饲养过程中存在喂食β-受体激动剂类物质情况,仅1批猪肉样品中苯乙醇胺A含量较高,个别肌肉及肾脏组织中存在受体激动剂多残留现象,未检测到克伦特罗、妥布特罗、西马特罗、苯氧丙酚胺和马喷特罗的存在。图1显示了实际样品中检测到11种受体激动剂残留的选择离子流图。
2.4 指紋特征研究
图2a为空白样品图谱,图2b、2c、2d分别为猪肌肉、猪心样品和有受体激动剂检出的猪心样品的总离子流图谱。研究发现,相同样品前处理条件与采集分析过程,动物的肌肉样品与肾脏样品谱图完全不同,在猪心样品5.0 min出现一未知物高峰,而对于肌肉样品是不存在的,如图2b、2c所示。因此,对于未知样品可利用样品谱图的相似性明确判定动物食品的组织部位,从而更有效的监测受体激动剂的残留部位。通过图2c和2d指纹谱对比可清晰确认受体激动剂组分的存在,例如,图2d中3.15 min、3.02 min出现两个峰对应物质为沙丁胺醇和特布他林。该指纹特征能够为动物源性食品中受体激动剂检测明确判定提供重要参考价值,
2.5 食品安全膳食评估
2.5.1 受体激动剂膳食暴露量评估依据
本研究根据所测得食物样品中β-受体激动剂污染水平和居民的食物消费量数据计算得到每人每日膳食暴露量[15-16]。计算公式为:
式中:D-每人每日膳食暴露量,?g/(kg bw·d);C-某类食物中β-受体激动剂含量,?g/kg;F-某类食物的消费量,g/d;W-体重,kg,体重按《2002年中国居民营养与健康状况调查报告》中的中国标准人体重63 kg计。
将获得的每人每日膳食暴露结果与食品添加剂联合专家委员会(JECFA)进行的风险评估结果得出的每日允许摄入量(ADI)值进行比较,得出暴露量占ADI值的百分比,安全性分析计算为:
占ADI值=D/ADI×100%
2.5.2 受体激动剂理论最大暴露量评估
以JECFA采用的动物性食品每日允许摄入量计算克伦特罗、沙丁胺醇和莱克多巴胺的理论暴露量。由于我国β-受体激动剂类药物如莱克多巴胺、克伦特罗、沙丁胺醇等在所有动物食品中不得检出,在屠宰环节莱克多巴胺、克伦特罗、沙丁胺醇等允许检出为0.50 ?g/kg。因此,本研究风险暴露评估以克伦特罗和沙丁胺醇ADI值0.004 ?g/(kg bw),莱克多巴胺ADI值1 ?g/(kg bw)为基准,以屠宰环节受体激动剂克伦特罗、莱克多巴胺、沙丁胺醇为例,对理论暴露污染物进行评估。屠宰环节对肌肉及脂肪未明确规定视为不得检出,本研究假设对肌肉、脂肪按方法最低检出浓度的1/2进行处理取值,即0.25 ?g/kg,未检出样品也按照方法最低检出浓度的1/2进行处理取值,理论暴露量计算结果见表3。
由表3可知,动物性食品中克伦特罗、莱克多巴胺和沙丁胺醇的总理论暴露量均小于相应的ADI值,分别占ADI值的63.44%、23.83%和59.54%,人体食入处于最大残留限量水平的食品的暴露量均低于ADI值,相对比较安全。由此可见,食用残留水平超过最大残留限值并不意味着一定对消费者有直接的健康风险,仅当每日膳食摄入量超过ADI值时会对消费者带来健康风险。
2.6 受体激动剂膳食暴露评估
欧洲食品安全局(EFSA)提出对具有遗传毒性和致癌性的物质的风险描述采用暴露边界值(Margin of Exposure,MOE)法,MOE是对动物或者人导致很小但可衡量作用的剂量与人或者动物的暴露量的比值。若物质导致不良作用的剂量与人群的摄入量越接近,即MOE值越低,表明对人群健康的危害越大。MOE按照公式计算如下:
MOE=BMDL10/D(2)
式中:MOE-暴露边界值;BMDL10-某种物质使10%的受试动物发生不良反应的剂量(统计数据),?g/(kg bw·d);D-某有害物质暴露量,/(kg bw·d)。
由于目前尚没有一个国际通用标准用来判定MOE值达到何种水平方表明危害物质的膳食暴露不对人体产生显著健康风险,风险评估结果的不确定性主要来源于数据资料的局限性和对毒理学、流行病学研究得到的客观资料分析和解释。因此,本研究BMDL10值拟采用克伦特罗每日容许摄入量ADI值0.004 ?g/(kg bw)来计算(1996年食品添加剂专家联合委员会第四十七会议提出);莱克多巴胺BMDL10值拟采用莱克多巴胺每日容许摄入量ADI值1 ?g/(kg bw)来计算(2006 年食品添加剂专家联合委员会第六十六届会议);其他9种受体激动剂BMDL10值拟参考克伦特罗的每日容许摄入量ADI值0.004 ?g/(kg bw)来计算。
本研究共检测动物食品肉类样本261批,检出11种受体激动剂残留21批,检出率8.05%,检出的11种受体激动剂含量范围为0.17~36.23 ?g/kg,根据联合国粮农组织数据中国人均每天肉类食品消费量(2004年)155.46 g,假设内脏类食品消费量为肉类的1/2,即77.73 g,结合检测结果评估动物肌肉及内脏食品中受体激动剂对人体健康的风险,结果见表4。
由表4可知,MOE值在0.031~6.450,随着MOE值的增加,暴露风险降低,前期研究发现,同一动物组织可能同时存在多种受体激动剂的情况,总暴露量MOE值低至0.02,暴露风险较高。按照国际食品法典委员会克伦特罗ADI值为0.004 ?g/(kg bw),则动物食品中受体激动剂测定总含量控制在1.08 ?g/kg对于人体健康相对安全。当动物组织中受体激动剂含量高于1.08 ?g/kg时,持续摄入可能具有较大的健康风险。因此为降低健康风险,建议消费者购买经检验检疫合格的动物食品。
3 结论
本研究采用固相萃取前处理,通过超高效液相色谱-串联质谱技术建立了动物源性食品中16种β-受体激动剂残留高灵敏、高选择检测方法。结果表明,在动物源性食品中检测到多种受体激动剂残留,进而确定动物饲养过程中可能存在喂食β-受体激动剂类药物情况。进一步根据研究结果进行膳食暴露评估,表明持续摄入含受体激动剂动物食品将增加健康风险。本研究为加强生猪养殖、屠宰过程中违法使用不同种类“瘦肉精”的监管提供了可靠的技术支撑,也可为动物源性食品中兽药残留、激素残留监控提供崭新的思路和方法,可实现食品安全问题的早发现、早处理,全面提高动物源性食品中药物残留的安全监管能力。
参考文献
[1]路平,肖肖,張衍海,等.我国“瘦肉精”监管现状分析及对策建议[J].中国动物检疫,2011,28(4):4-6.
[2]栗艳.“瘦肉精”的危害及防治[J].中国畜牧兽医文摘,2011,27(4):175.
[3]Simon T,Shellaiah M,Steffi P,et al. Development of extremely stable dual functionalized gold nanoparticles for effective colorimetric detection of clenbuterol and ractopamine in human urine samples[J].Analytica Chimica Acta, 2018,1023:96-104.
[4]SAI F,HONG M,Y F ZHAO,et al.Simultaneous detection of residues of 25 β2-agonists and 23 β-blockers in animal foods by high-performance liquid chromatography coupled with linear ion trap mass spectrometry[J].Journal of Agricultural and Food Chemistry,2012,60:1898-1905.
[5]GUO P,WAN J C,ZHAN C R,et al.A simplified sample pretreatment for the rapid determination of 22 β-agonist residues in swine muscle and liver tissues by ultra-high-performance liquid chromatography tandem mass spectrometry[J].Journal of Chromatography B,2018,1096:122-134.
[6]CHANG K C,CHANG Y T,TSAI C E.Determination of ractopamine and salbutamol in pig hair by liquid chromatography tandem mass spectrometry[J].Journal of Food and Drug Analysis,2018,26:725-730.
[7]SILVAGON V C ?ALVESKELLY, SANTOS T J.Optimization of a multiresidue and multiclass analysis method for anabolic agents and β2-agonists in bovine urine by GC-MS/MS[J].Microchemical Journal,2017,133:551-555.
[8]LU M H,ZHANG L,LI X,et al.A new method for the analysis of β2-agonists in human urine by pressure-assisted capillary electrochromatography coupled with electrospray ionization-mass spectrometry using a silica-based monolithic column[J].Talanta,2010,8:1655-1661.
[9]QIU X Z,XU X Y,LIANG Y,et al.Fabrication of a molecularly imprinted polymer immobilized membrane with nanopores and its application in determination of β2-agonists in pork samples[J].Journal of Chromatography A,2016,1429:79-85.
[10]DONG B L,ZHAO S J,LI H F,et al.Design, synthesis and characterization of tracers and development of a fluorescence polarization immunoassay for the rapid detection of ractopamine in pork[J].Food Chemistry,2019,271:9-17.
[11]GAO F,WU M L,ZHANG Y,et al.Sensitive determination of four β2-agonists in pig feed by capillary electrophoresis using on-line sample preconcentration with contactless conductivity detection[J].Journal of Chromatography B,2014,973:29-32.
[12]MUTHUMARIYAPPAN A,RAJAJIA U,CHEN S M,et al.Sonochemical synthesis of perovskite-type barium titanate nanoparticles decorated on reduced graphene oxide nanosheets as an effective electrode material for the rapid determination of ractopamine in meat samples[J].Ultrasonics Sonochemistry,2019,56:318-326.
[13]ZHANG L H,WANG Q W,QI Y,et al.An ultrasensitive sensor based on polyoxometalate and zirconium dioxide nanocomposites hybrids material for simultaneous detection of toxic clenbuterol and ractopamine[J].Sensors and Actuators B:Chemical,2019,288:347-355.
[14]Xiao X H,Yan K L,Xu X F,et al.Rapid analysis of ractopamine in pig tissues by dummy-template imprinted solid-phase extraction coupling with surface- enhanced Raman spectroscopy[J].Talanta,2015,138:40-45.
[15]苗紅.食品及生物材料中β-激动剂和β-阻断剂残留检测技术研究及污染评价[D].北京:中国疾病预防控制中心,2010.
[16]刘畅.食品中兽药残留高通量筛查与检测平台的建立及膳食暴露评估研究[D].上海:第二军医大学,2013.
