遗传性釉质发育不全相关基因FAM83H 突变的初步研究
2022-03-07胡仲琳刘伟才
胡仲琳, 刘伟才
(上海牙组织修复与再生工程技术研究中心,同济大学口腔医学院,同济大学附属口腔医院口腔修复教研室,上海 200072)
遗传性釉质发育不全(amelogenesis imperfecta,AI)是一组相似疾病的总称,是指在牙齿发育过程中, 由于遗传因素而引起的牙釉质形成或矿化障碍,不伴有其他全身性疾病,可同时伴有前牙开牙合或牙齿敏感症等[1]。根据调查人群的不同,AI 的患病率为1/14 000~1/12 000,是口腔科学领域的遗传性疾病。 AI 发病原因及其致病因素众多,并且临床表现也有很大不同,具有临床异质性和遗传异质性[2]。AI 患者临床表现包括牙釉质的厚度、硬度及颜色等的异常,主要分为4 类:发育不全型、钙化不全型、成熟不全型及复合型[3]。 AI 的遗传方式有常染色体显性遗传、 常染色体隐性遗传及X 性连锁遗传,目前研究者已经证实多个与AI 相关的致病基因,如釉原蛋白基因 (amelogenin,AMELX)、 釉蛋白基因(enamelin,ENAM)、激肽释放酶(kallikrein-4,KLK-4)编码基因等[1,4]。
FAM83H 属于FAM83 家族, 位于染色体8q24.3,包含5 个外显子,由1 179 个氨基酸组成,绝大部分氨基酸由五号外显子编码。该基因在2008年被首次报道与常染色体显性釉质钙化不全型AI相关, 目前所知道的致钙化不全型AI 的FAM83H基因突变众多,致病性突变通常位于最后一个外显子中编码287 位丝氨酸至694 位谷氨酸的序列之间[5-7]。 目前,关于FAM83H 突变导致钙化不全型AI的发病机制尚不清楚,本课题前期研究[8-9]发现,一个FAM83H 致病突变(c.1354C>T, p.Gln452*)就能导致钙化不全型AI。为进一步理解AI 的发病机制,本研究利用细胞功能实验对FAM83H 第452 位谷氨酰胺位点突变的致病性进行验证。
1 材料和方法
1.1 材料和仪器
DMEM 高糖培养基,胰酶消化液,青/链霉素混合液(HyClone 公司,美国);胎牛血清(Gibco 公司,美国);TRIzol 试剂盒(Invitrogen 公司, 美国);FAM83H 野生型及突变型病毒表达载体由上海创济生物科技有限公司(中国)设计合成;DAPI 溶液(Solarbio 公司,中国);PCR 工作仪(Bio-red 公司,美国);荧光定量PCR 仪(Roche 公司,德国);荧光显微镜(Nikon 公司,日本)等。
1.2 实验方法
1.2.1 细胞培养 采用含10%胎牛血清的DMEM培养基培养永生化人胚胎肾上皮细胞HEK293T,并将其置于37 ℃、5%CO2的细胞培养箱内培养,1~2 d传代1 次。
1.2.2 稳转细胞系构建 将HEK293T 细胞铺在皿中,待细胞密度长至70%~80%后,分别转染pCDH慢病毒空载、pCDH-FAM83H-wild-HA 慢病毒表达载体及pCDH-FAM83H-mut-HA 慢病毒表达载体,Sanger 测序进行验证。 转染24 h 后,采用嘌呤霉素(puromycin)对细胞株进行筛选,筛选10 d 后,荧光显微镜下观察转染效率。
1.2.3 RNA 提取与实时定量聚合酶链反应(RTqPCR) 上述3 组细胞稳转株以1×106/孔的密度接种于含有适量完全培养基的12 孔细胞培养板中,待细胞密度至80%~90%后, 将实验细胞用磷酸缓冲盐溶液(phosphate buffer saline,PBS)冲洗3 次,使用TRIzol 法提取细胞总RNA,测量其浓度;根据TaKaRa 公司逆转录试剂盒说明书操作,将RNA 转换为cDNA;按照RT-qPCR 试剂说明书,进行操作。FAM83H 正向引物序列:5'-GGCCCCTCACATTTGGCTT-3',反向引物序列:5'-GCAGATCCACGTCGGTAAACA-3';内参对照GAPDH 正向引物序列:5'-CATCTGAGGGCCCACTG-3',反向引物序列:5'-GAGGCCATGTAGGCCATGA-3'。 反应程序为30 s,95 ℃;5 s,95 ℃;30 s,60 ℃,共45 个循环。
1.2.4 免疫荧光 将灭菌处理的细胞爬片放入24 孔板中,以6×103/孔的密度将FAM83H-wild 和FAM83H-mut 细胞种于细胞爬片上。 细胞汇合达80%时,弃去培养基,PBS 溶液漂洗3 次,用4%多聚甲醛室温固定20 min;PBS 漂洗3 次,加入含DAPI 的荧光封片剂封片,固定后于荧光显微镜下观察。
1.2.5 统计学分析 采用GraphPad prism 8.0 软件对基因间的数据进行处理。 采用t检验法比较2 组之间的差异,P<0.05 表示差异具有统计学意义。
2 结果
2.1 病毒表达载体构建
课题组前期文献及家系研究[8-9]发现,位于人类染色体Chr8q24.3 区域FAM83H 基因第5 号外显子的第1354 位发生C>T 的杂合突变 (c.1354C>T, p.Gln452*),该突变导致钙化不全型AI,且该位点氨基酸谷氨酰胺在小鼠、大鼠、狼、黑猩猩等物种中具有高度保守性,说明该位点对于维持FAM83H 基因的功能有重要作用。根据基因突变序列构建突变病毒载体,以期检测该位点突变后该蛋白对细胞的功能影响,对病毒载体进行测序确认载体构建成功(图1)。

图1 病毒表达载体测序结果Figure 1 Sequencing results of viral expression vector
2.2 载体转染效率检测
pCDH 慢病毒空载、pCDH-FAM83H-wild-HA 及pCDH-FAM83H-mut-HA 慢病毒表达载体分别感染对数期生长的293T 细胞,感染48~72 h 后,细胞高表达绿色荧光蛋白(图2)。
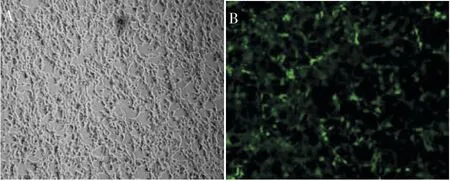
图2 转染后48 h 的细胞荧光图(×40)Figure 2 Fluorescence picture of cells 48 h after transfection(×40)
2.3 RT-qPCR 检测
RT-qPCR 结果显示,转染后,与野生型相比,突变型FAM83H mRNA 表达水平降低(图3)。

图3 RT-qPCR 结果Figure 3 RT-qPCR results
2.4 细胞荧光实验
FAM83H 蛋白免疫荧光发现, 野生型FAM83H蛋白主要在细胞质内, 而突变后的FAM83H 蛋白进入细胞核,这说明突变型FAM83H 导致AI 的主要机制可能为在细胞内的作用位点不同所导致的(图4)。
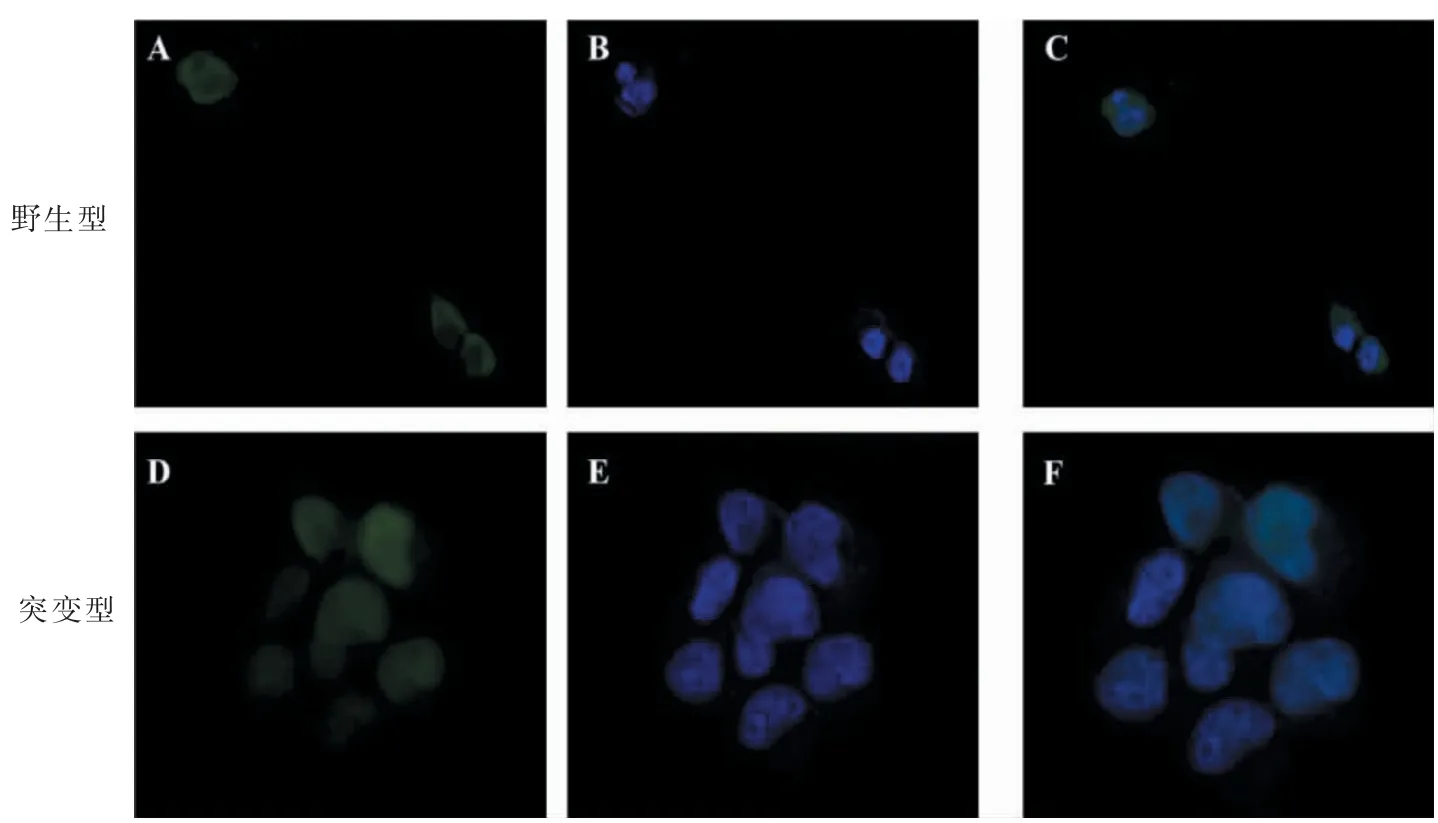
图4 2 组免疫荧光染色结果(×400)Figure 4 Immunofluorescence results of two groups(×400)
3 讨论
本研究成功构建了pCDH-FAM83H-wild-HA 慢病毒表达载体及pCDH-FAM83H-mut-HA 慢病毒表达载体。转染后,293T 细胞形态无明显变化,转染效率高,并且成功构建了293T 稳转细胞株。 FAM83H基因突变研究结果发现, 相较于野生型, 突变型FAM83H 的mRNA 表达水平降低;细胞荧光染色结果显示,突变后的FAM83H 蛋白主要作用在细胞核内,而野生型FAM83H 蛋白主要在胞质中。
FAM83H 是一个胞内蛋白,主要分布于高尔基体,因此推测其功能与高尔基体密切相关。FAM83H基因编码非特异性胞内蛋白,其除在牙齿发育过程表达外,还在肾、膀胱、喉及子宫等多种组织中表达[10]。Kim 等[11]在2008 年首次报道FAM83H 为钙化不全型AI 的主要致病基因, 随后越来越多的FAM83H 基因突变位点被发现,多数致病性突变位点均位于5 号外显子287 位至694 位氨基酸之间。Kweon 等[12]和Wang 等[13]构 建FAM83H 过 表 达 和 敲除转基因小鼠模型,发现小鼠釉质组织学形态无明显变化,表明单纯FAM83H 功能亢进或丧失可能不是导致AI 的主要致病机制。 目前比较认同的说法是,FAM83H 致病性突变可能导致蛋白作用区域发生变化, 同时细胞免疫荧光结果显示, 突变后的FAM83H 蛋白主要表达于细胞核中。 尽管FAM83H突变可逃避无义介导的mRNA 降解,有学者发现位于470 谷氨酰胺至610 位谷氨酸之间较少有无义突变的发生,可能与该区段作用于无义介导的mRNA降解相关,该区段无义突变后无法转录翻译,导致FAM83H 功能丧失, 患者临床釉质发育形态无异常,导致该区段突变报道较少[7]。 但实验结果发现突变后mRNA 表达降低,可能是突变后转录水平下降的原因[6]。 FAM83 蛋白家族的成员通过其共有的N端DUF1669 结构域与异位丝氨酸-苏氨酸蛋白激酶CK1 家族的亚细胞相互作用并调节其亚细胞分布,FAM83H 与CK1 亚型共定位于细胞质和细胞核形成斑点状结构。Kuga 等[14]发现FAM83H 通过氨基端与CK1α 结合,羧基端与角蛋白丝结合,招募CK1α到角蛋白丝中调节角蛋白细胞骨架的形成;FAM83H 过表达的癌细胞中还有上皮细胞极性的缺失和E-钙黏蛋白的表达, 提示FAM83H 过表达诱导的CK1α 介导的角蛋白丝拆卸参与了结直肠癌的侵袭和/或转移。 同时相关研究表明,突变后的FAM83H 携同CK1α 入核,导致胞质内β-连环蛋白(β-catenin)磷酸化减少,转位入核增多,核积聚现象发生,致使Wnt/β-catenin 信号通路激活,抑制釉质矿化[15]。 此外,肝癌组织标本中FAM83H 的核表达与较高的肿瘤分期和较高的术前血清α-甲胎蛋白水平有显著关系,FAM83H 的表达升高对肝癌患者治疗效果及预后有着重要影响,同时原癌基因MYC参与了FAM83H 表达的转录调节[16]。 有研究表明,FAM83H 与NCK1/2 酪氨酸激酶相互作用和共定位,这种相互作用由FAM83H 末端富含脯氨酸的基序介导,它们与NCK1/2 的第2 个和第3 个SH3 结构域特异性地相互作用;而触发FAM83H C 端截短的致病性AI 突变蛋白保留了它们与CK1 亚型的相互作用,但与NCK1/2 失去了相互作用,其相互作用机制及影响尚不清楚[17]。 尽管对于釉质发育不全致病基因FAM83H 的研究越来越多,然而其在釉质形成过程中的机制尚不明确,需进一步探索FAM83H在釉质发育不全中的作用及致病机制,为釉质发育不全疾病的防治提供可靠依据。
综上所述,本文通过构建FAM83H 基因突变表达载体,对转染后的293T 细胞进行RT-qPCR 及细胞免疫荧光检测,分析突变后FAM83H 基本功能的变化,证明该位点突变后FAM83H 蛋白在细胞内作用位点发生了改变,FAM83H 在调控釉质形成的过程中,可能对多种不同的信号通路均有影响。 近期的研究表明,JNK 和p38 可能是FAM83H 表达的关键调控元件,JNK/p38 MAPK 通路在NaF 诱导的FAM83H 表达中起着至关重要的作用[18];FAM83H通过调节CK1α 来调节经典Wnt/β-catenin 信号通路。这为我们发掘FAM83H 调控钙化不全型釉质发育不全症机制,提供了有力依据。 为了更好地利用FAM83H 在釉质发生、发展过程中的作用,促进釉质发育不全生物治疗的发展,有关FAM83H 在釉质形成过程中的作用,以及其突变后如何影响釉质形成,这些方面需要进一步探索。
