1个结蛋白基因新突变致扩张型心肌病家系的临床特点及遗传分析
2022-03-05于莉宋贵波靳冰玉干学东邱雪平王陈程亚婷郑芳
于莉,宋贵波,靳冰玉,干学东,邱雪平,王陈,程亚婷,郑芳
(1.武汉大学中南医院 a.基因诊断中心&检验科 b.心血管内科,武汉 430071;2.昆明医科大学第一附属医院检验科,昆明 650032)
扩张型心肌病(dilated cardiomyopathy, DCM)简称扩心病,是一种以心室(左室或双室)收缩功能受损及心室扩张为主要特征的常见心肌病,其发病率约为1∶250~1∶2 500,可进展为心力衰竭,是世界范围内心脏移植最常见的适应证[1]。研究表明,高达40%的患者由遗传因素致病,除常见致病基因如肌联蛋白基因(titin,TTN)、肌球蛋白重链基因(myosin heavy chain 7,MYH7)和核纤层蛋白A/C基因(lamin A/C,LMNA)等外,约有1%~2%的DCM患者与结蛋白(desmin,DES)基因突变有关[2-3]。DES基因编码中间丝(intermediate filament, IF)蛋白质中的结蛋白,其特异性表达于肌细胞,在心脏的生长发育中起重要作用,该基因突变后可能会影响中间丝正常的组装、结构及功能,从而诱发心肌病和骨骼肌病[3]。本研究拟通过全外显子组测序、Sanger测序等方法鉴定1个新的致遗传性扩张型心肌病的DES基因变异,以期为家系遗传咨询提供试验依据。
1 资料和方法
1.1一般资料 先证者(Ⅱ-9),男,52岁,曾于2013年至2021年在武汉大学中南医院就诊,临床确诊为扩张型心肌病,在39岁时因房室传导阻滞行心脏起搏器植入术,目前已发展为心力衰竭,同时气促伴双下肢无力。该扩张型心肌病的家系图谱见图1。

注:*,采集过外周血标本的个体。
先证者自述其父亲(Ⅰ-1)为健康人,89岁去世,且兄弟姐妹未曾出现相关表型,其母亲(Ⅰ-2)在59岁时因心脏疾病去世,同时母亲的妹妹也患有心脏疾病。该家系的另外4名成员(Ⅱ-1、Ⅱ-5、Ⅱ-11及Ⅲ-1)也被确诊为扩张型心肌病伴房室传导阻滞。Ⅱ-1个体于52岁去世,曾因房室传导阻滞进行过2次心脏起搏器植入术。其余3名成员Ⅱ-5、Ⅱ-11及Ⅲ-1也都进行了心脏起搏器植入术,最终发展为心力衰竭需进行心脏移植治疗。此外,Ⅲ-1个体发病早,26岁时就植入心脏起搏器,目前同样出现气促和下肢无力的情况。该家系患者的心电图和超声心动图结果见表1。
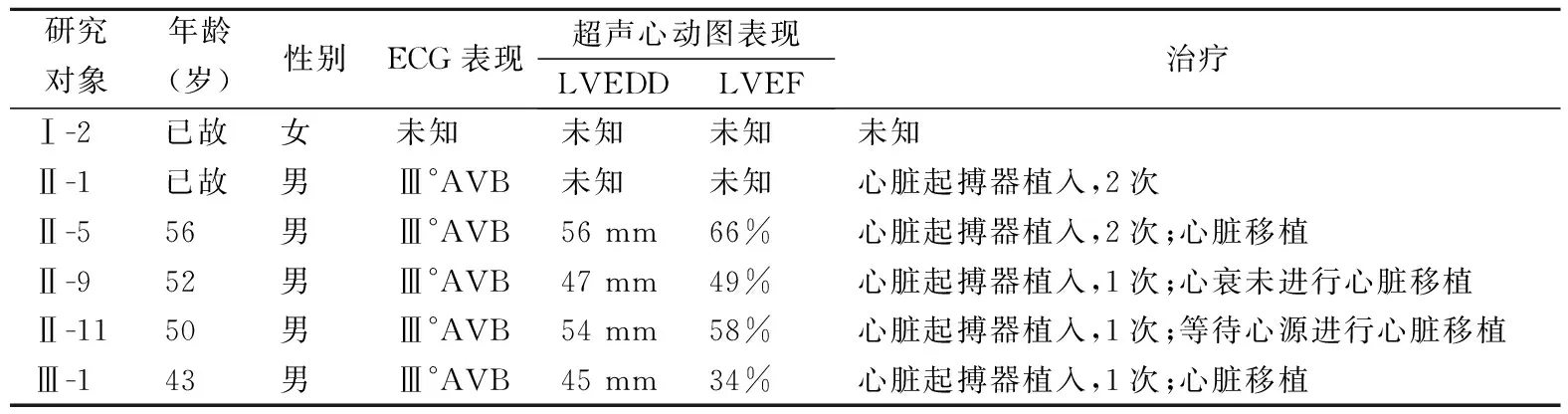
表1 扩张型心肌病家系患者临床资料
1.2方法
1.2.1主要仪器及试剂 普通PCR仪及核酸电泳仪(美国Bio-Rad公司),ABI 3730s型DNA序列自动分析仪(美国ABI公司),NanoDrop one超微量紫外分光光度计(美国Thermo Scientific公司)。PCR扩增试剂2×Taq Plus Master Mix(南京诺唯赞生物科技股份有限公司),外周血DNA提取试剂盒(Quick-DNA Miniprep Plus Kit,美国Zymo Research公司)。
1.2.2外周血采集及DNA提取 采集该家系成员就诊时的静脉血(EDTA-K2抗凝)2 mL,采集后立即按照外周血DNA提取试剂盒说明书提取DNA样本。DNA样本的纯度和浓度用NanoDrop one超微量紫外分光光度计测定,选取纯度(A260 nm/A280 nm为1.7~1.9,A260 nm/A230 nm≥2.0)及浓度较高的样本,置于-20 ℃保存。
1.2.3全外显子组测序及可疑致病基因筛选 全外显子组测序委托深圳华大临床检验中心进行,通过打断DNA制备文库,用Roche KAPA HyperExome探针捕获和富集目标基因外显子及邻近剪切区,最后使用MGISEQ-2000测序平台进行检测,质控为目标区域平均测序深度≥180×,其中目标区平均深度>20×的位点占比>95%。测序数据分析通过BWA(Burrows-Wheeler Aligner)软件与UCSC数据库提供的hg19版的人类参考基因组进行比对,去除重复。然后使用GATK(Genome Analysis Toolkit)工具包寻找变异。最后通过在多个人群和疾病数据库(如千人数据库、HGMD数据库、ESP数据库等)中比对、生物信息工具注释,根据位点的数据库频率筛选可能的致病性位点,根据ACMG制定的变异分类标准与指南[4]评分进行变异致病性分类,即可得到与扩张型心肌病相关的可疑致病位点。最后筛选出符合先证者家系遗传模式及发病特点的可疑致病基因突变位点进行Sanger测序及变异致病性分析验证。
1.2.4Sanger测序验证 针对上述分析获得的可疑致病变异位点DESc.140G>T(p.Ser47Ile),用在线引物设计工具NCBI Primer-BLAST设计突变位点的引物,并交由武汉擎科生物技术有限公司合成。引物序列涵盖DES基因(NC_000002.12)的该变异位点。正向引物序列(5′→3′):CCAGGCCTACTCGTCCAGC,反向引物序列(5′→3′):CTCCTCCTCGTAGAGCTCGG,产物片段长度为469 bp,退火温度为60 ℃。普通PCR扩增体系为20 μL,包括:2×Taq Plus Master Mix(Vazyme biotech,P211)10 μL,正、反向引物(浓度为10 μmol/L)各1 μL,DNA模板2 μL,ddH2O 6 μL。循环参数:95 ℃预变性5 min;95 ℃变性30 s,60 ℃退火30 s,72 ℃延伸35 s,循环35次;72 ℃延伸10 min;4 ℃低温保存。
PCR结束后,用10 g/L琼脂糖凝胶电泳验证扩增效果,将469 bp片段大小的扩增产物送至武汉擎科生物技术有限公司,用ABI 3730s型DNA序列自动分析仪直接测序分析。采用Chromas软件读取并比对测序结果以验证突变位点序列。
1.2.5变异致病性分析 针对DESc.140G>T(p.Ser47Ile)变异位点,用在线工具UniProt、PolyPhen-2和Mutation taster对该位点的保守性和致病效应进行确认。随后用ANTHEPROT 6.9.3软件分析蛋白质二级结构及理化性质(亲疏水性、抗原性等)的改变,并用PyMOL软件对蛋白质三级结构的影响进行分析。最后用PhosphoSitePlus数据库(https://www.phosphosite.org/homeAction)对突变造成蛋白质翻译后修饰的改变进行预测。
2 结果
2.1全外显子组测序分析 该家系的全外显子组测序结果提示共存在十余种基因变异位点,其中与扩张型心肌病有关的基因有桥粒芯蛋白2基因(desmoglein 2,DSG2)、PRDM16基因(PR/SET domain 16,PRDM16)、DES基因。然而在该家系中与疾病共分离的只有DES基因突变,同时该突变的致病特点也符合该家系疾病的常染色体显性遗传模式(图1)。故最终筛选出1个位于2号染色体DES基因的杂合错义突变c.140G>T(p.Ser47Ile)。
2.2Sanger测序结果 先证者(Ⅱ-9)及家系其他已发病患者(Ⅱ-5、Ⅱ-9、Ⅱ-11、Ⅲ-1)DES基因第一外显子均存在c.140G>T(p.Ser47Ile)的杂合突变。而健康的家系成员Ⅱ-3、Ⅱ-7、Ⅲ-3、Ⅲ-5、Ⅲ-8均为野生型(图2)。

注:A,携带杂合突变成员的Sanger测序结果;B,健康个体的Sanger测序结果;红色箭头所指即为突变位点。
2.3变异位点致病性分析 生物信息学方法的验证与预测结果显示,在人、猩猩、小鼠、大鼠、牛等多个物种中该位点高度保守(图3);而PolyPhen-2和Mutation taster均提示该突变为有害突变(图4A);该突变会造成蛋白质二级结构的改变(图4B),蛋白质疏水性增强和抗原性下降(图4C),蛋白质内部氢键缺失(图4D);该变色位点的丝氨酸是潜在的磷酸化位点,突变可能会影响蛋白质磷酸化状态的改变(图4E)。故而该变异位点满足PP3证据项(当用多种生物信息学软件进行预测时,均提示该突变有害)。此外,根据家系Sanger测序结果,该变异位点在家系中满足共分离(PP1证据项)。该家系变异携带者的表型和家族史高度符合某种单基因遗传疾病(PP4证据项)。

注:箭头处为人结蛋白头部第47位丝氨酸,显示该位点在多种物种中高度保守。
该DCM家系的DES基因发生杂合错义突变c.140G>T(p.Ser47Ile),遗传模式为常染色体显性遗传。在全外显子组测序报告中,该变异位点被解读为意义未明的变异(PM1:该变异位于热点突变区域+PM2:在千人基因组、ESP数据库和gnomAD数据库中均未被收录)。但当对该家系的疾病共分离以及突变功能预测的生物信息学等情况进行分析后,发现该变异可升级为“可能致病的”变异(PM1+PM2+PP1+PP3+PP4)。

注:A,Mutation taster和Polyphen-2预测结果;B,蛋白质二级结构预测结果,突变位点红线标记,野生型(上)突变型(下);C,理化性质预测结果对比图,突变位点红线标记,野生型(左)突变型(右),3条曲线依次代表疏水性、抗原性和亲水性;D,蛋白质三级结构的预测,白框部分显示突变型第47位氨基酸氢键消失;E,PhosphoSitePlus数据库中对人结蛋白潜在的磷酸化位点预测图,箭头处为突变位点。
3 讨论
本研究涉及一遗传性扩张型心肌病家系的基因诊断,该家系的致病突变DESc.140G>T(p.Ser47Ile)出现在染色体2q35上DES基因第一外显子区域,导致第47位的丝氨酸被异亮氨酸取代,可能使蛋白质的二、三级结构以及蛋白质的疏水性和抗原性等理化性质发生改变,甚至可能影响蛋白质的磷酸化状态。
DES基因编码一种特异性表达于肌细胞的Ⅲ型中间丝蛋白即结蛋白,对于维持心脏正常的结构、传导及心肌收缩等功能非常重要,同时也参与细胞器定位组装、细胞机械应力传递、细胞信号传导等过程[3,5]。因此,当DES基因发生突变,就可能导致结蛋白结构及中间丝网络的异常,影响上述生物过程的正常进行,最终导致心肌病和骨骼肌病,又称为结蛋白相关肌病(desmin-related myopathies,DRM)[6]。
结蛋白的结构分为两侧非螺旋的头尾部及中间的螺旋杆状区域,本研究涉及的突变发生在结蛋白的头部结构域,结蛋白的头部结构域参与自身的组装过程,与尾部相互作用可以保护结蛋白的氨基末端免受蛋白酶的分解从而维持稳定,同时由于在结蛋白头部丝氨酸的数量占比超过1/5,故而存在多个潜在的磷酸化修饰位点,可参与蛋白质翻译后的修饰。而本研究报道的DESc.140G>T(p.Ser47Ile)突变使原来的丝氨酸被异亮氨酸取代,疏水性增强的同时或可改变结蛋白的磷酸化状态。笔者发现的突变与其他报道的致病突变具有一定共性,如第6、7、12、13、28、46位丝氨酸同样被预测为潜在的磷酸化位点[1,7],其致病性突变也都是被疏水性氨基酸所取代,当患者出现心脏表型时皆表现出DCM同时都伴有包括房室传导阻滞(atrioventricular block,AVB)在内的心律失常,且除心肌外其他肌肉症状较轻。此外,这些突变体的组织病理学结果较为相似,均显示中间丝网络的损坏、异常结蛋白胞浆内聚集、线粒体和溶酶体结构功能异常等,DESc.140G>T(p.Ser47Ile)突变导致该家系扩张型心肌病的病理机制可能与之相似。
DES基因突变的临床表型与DCM常见的MYH7致病基因突变类似,均具有较大的异质性[8],可导致除DCM外的多种心肌病和(或)近远端肌病,甚至影响面部、呼吸和消化功能等,推测DRM的异质性可能与心肌骨骼肌结构及再生能力的不同有关。而DES基因突变致心肌病的同时往往伴随传导阻滞等问题,考虑因结蛋白在浦肯野纤维中含量丰富[3],突变会影响心脏正常的传导功能。本研究中,患者发病年龄总体较对照提前,但幼年不发病,首先出现房室传导阻滞,随后发展为DCM伴Ⅲ°AVB,最终结局为心衰。如Ⅲ-1个体在26岁时就已植入心脏起搏器,而Ⅲ-7和Ⅳ-1个体虽携带DESc.140G>T(p.Ser47Ile)突变但目前尚未发病,考虑和年纪较小有关。此外,该家系部分成员随年龄增长出现下肢无力的症状,但无法判断是由心衰造成的还是突变导致的肌肉改变。
虽然已经报道的DES致病性突变已有100多种,但DRM具体的致病机制尚不清晰。有研究认为DRM随年龄进展的原因是肌细胞中间丝网络受损,机械应力损伤增加及线粒体功能的紊乱,而不是结蛋白的聚集[9]。本研究中该家系患者主要出现的是心脏表型,有1~2例心脏移植的患者,然而手术均是在外院完成难以获得其心脏组织,故未能进行病理机制及组织病理学方面的验证,这也是本研究的不足之处,后续可通过构建细胞或动物模型对其机制进行深入研究。
本研究发现了DES基因的新突变c.140G>T(p.Ser47Ile),在一定程度上丰富了DES基因突变谱,也为遗传咨询提供数据支持。由于该突变还伴有心脏的传导阻滞,严重时有发生心脏骤停和猝死的风险,如果能进行分子诊断将有助于临床的早期确诊,以及在症状出现前确定合适的预防和管理策略。
