哈尔满碱分子光谱的密度泛函理论研究
2022-03-04陈玉锋关皓月邵长斌
陈玉锋,关皓月,邵长斌,姜 力
(牡丹江师范学院 化学化工学院,牡丹江 157011)
1 引 言
咔啉类化合物包含一个三环吡啶并(3,4-b)吲哚结构,β-咔啉类化合物中吡啶环中氮原子在2位,故哈尔满碱(Harmane)归属于β-咔啉类生物碱,β-咔啉类生物碱广泛分布于自然界的动植物中,由于β-咔啉类化合物具有广泛的药理和生理活性,包括抗癌、抗病毒、抗高血压、抗氧化等,因而被大量人工合成,并得到广泛应用[1-5].
Canamares等[6,7]使用DFT理论研究了几种哈尔满碱分子的拉曼光谱和表面增强拉曼光谱,并指出了这几种分子和Ag增强基底之间的吸附方式.
对于哈尔满碱分子红外光谱、振动方式的指认、前线轨道、激发态等微观结构的理论研究未见报道.本文采用密度泛函理论,M062X/6-31g(d)水平上计算了哈尔满碱分子的红外光谱,并和实验采集的红外光谱进行了对比,利用VEDA4软件对哈尔满碱分子的简正振动模式进行了指认归属[8],并结合TDDFT计算结果分析了哈尔满碱分子的吸收光谱和激发态.
2 理论计算与实验
首先采用Gaussian09量子化学程序包[9],M062X/6-31g(d)基组水平上对哈尔满碱分子的几何结构进行优化.并进行频率计算,IR做图时频率校正因子为0.947[10],前线分子轨道、振动归属分析和分子静电势分析借助于Gauss View 6.0程序完成.哈尔满碱分子购于aladdin化学试剂公司(样品纯度为95%),红外实验光谱采集采用赛默飞世尔公司的is10傅里叶红外光谱仪.
3 结果与讨论
3.1 分子结构优化
使用DFT理论M062X/6-31g(d)水平下对哈尔满碱分子进行结构优化,优化后的分子结构稳定,计算结果中未出现虚频,说明优化得到的哈尔满碱分子为稳定构型.分子结构如图1所示.
哈尔满碱分子由24个原子组成,优化结果表明除了甲基上的3个氢原子外哈尔满碱其余原子均在一个平面上,故哈尔满碱分子的结构为近平面的.哈尔满碱分子上的吡啶环中两个C-N的键长分别为0.1346 nm、0.1328 nm,而吡咯环中C-N的键长为0.1382 nm.
3.2 哈尔满碱的谱带振动与归属
表1列出了哈尔满碱分子振动模式中主要的特征峰的未矫正频率和矫正频率,以及红外振动强度和振动归属.图2中a为理论计算得到的红外光谱图、b为实验测得的哈尔满碱分子的红外光谱图,对400 cm-1至1800 cm-1频率范围内的特征峰进行了指认和归属,主要的振动模式有:1629 cm-1归属于C-C伸缩和C-H面内弯曲.在1577 cm-1归属于吡啶环上18号N原子与邻位C原子伸缩振动、C-C伸缩振动、C-C-C面内弯曲振动.1483 cm-1处归属于C-C伸缩振动、C-H面内摇摆振动和23号N原子与24号H原子面内弯曲振动.1441 cm-1处归属于C-C伸缩振动、H-C-C面内弯曲振动和甲基C-H键的剪式振动.在1405 cm-1处归属于N23-C8伸缩振动、C-H面内弯曲振动和C-C-N面内弯曲振动.1296 cm-1处归属于C-C伸缩振动、C-H面内弯曲振动和C-C-C面内弯曲振动.在1248 cm-1处归属于C-C伸缩振动、23号N原子与8号C原子伸缩振动.950 cm-1处归属于甲基的面外弯曲振动和H-C-C面内弯曲振动.在805 cm-1、761 cm-1、734 cm-1、721 cm-1处都归属于C-H的面外弯曲振动.在565 cm-1归属于C-C伸缩振动、C-C-C面内弯曲振动和C-C-N面内弯曲振动.
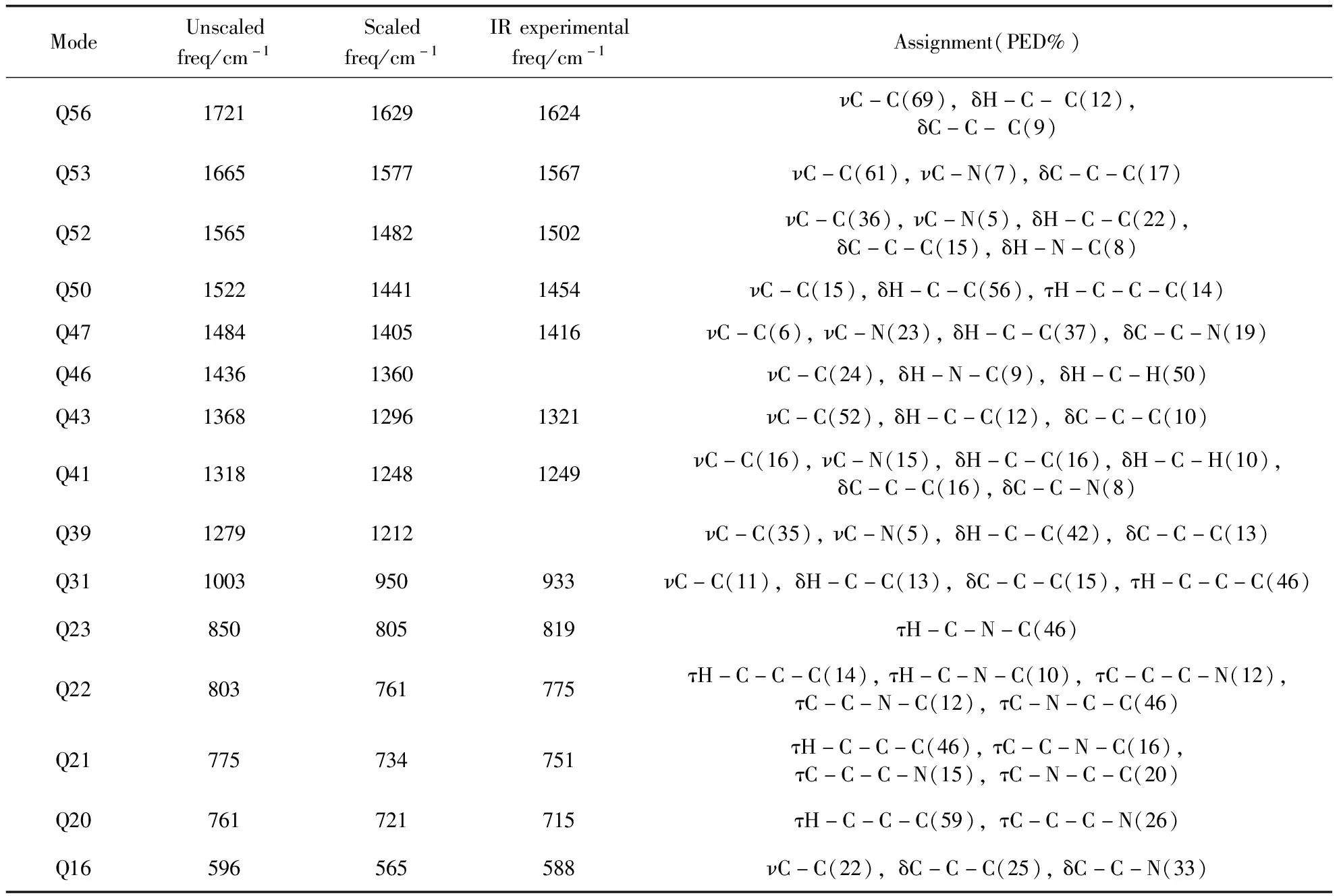
表1 哈尔满碱计算频率、振动归属
3.3 哈尔满碱分子的表面静电势
分子周围某点的表面静电势(Molecular electrostatic potential)是指从无穷远处的一个单位正电荷移动到该点时做的功.静电势对于考察分子间静电相互作用、预测分子凝聚相性质、预测反应位点等方面有重要意义[11,12].静电势是由体系电荷分布直接决定的,静电势图上不同颜色代表不同的电荷分布,静电势图蓝色区域代表负电性集中区域,容易吸附亲电试剂、红色区域为正电性集中区域,容易吸附亲核试剂.从哈尔满碱分子静电势图(图3)可以看,哈尔满碱分子有9个极大值点,极大值为24号H原子,极大值的能量为51.322 kcal/mol,有7个极小值点,极小值点的位置是18号N原子,极小值能量为-38.804 kcal/mol.
3.4 哈尔满碱分子的前线轨道分析
分子轨道及其性质、能量、前线电子密度对于物质的物理化学性质,预测化学反应的位点等都具有重要意义.分子轨道中的最高占据分子轨道(highest occupied molecular orbitals ,HOMOs)和最低未占据分子轨道(lowest unoccupied molecular orbital,LUMO)及其能级差常用来判断分子的稳定性、化学活性等性质[13,14].如图4所示为哈尔满碱分子的 HOMO、LUMO轨道,能级差为6.76 eV,HOMO-LUMO能级间电子跃迁的激发线波长λ=293.92 nm.
3.5 哈尔满碱分子吸收光谱和激发态
哈尔满碱分子的TDDFT激发态计算所得到的吸收光谱图如图5所示,a为在气相条件下哈尔满碱分子光谱理论计算的吸收波长为206.15 nm、b为以水为溶剂的哈尔满碱分子理论计算的吸收波长为222.88 nm、c为以乙醇为溶剂的哈尔满碱分子理论计算的吸收波长为223.06 nm,d为以四氯化碳为溶剂的哈尔满碱分子理论计算的吸收波长为223.27 nm,e为乙醇溶液实验获得的紫外吸收波长为292 nm.可以看出随着溶剂极性的降低,吸收波长发生红移.
4 结 论
采用密度泛函理论,M062X/6-31g(d)水平上对哈尔满碱分子进行了结构优化以及频率计算,绘制了哈尔满碱分子的红外光谱,和实验采集的红外光谱进行了比对,具有较好的一致性,结合势能函数分布,采用VEDA4程序对哈尔满碱分子的简正振动模式进行了指认和归属.绘制了分子的前线轨道,前线轨道能级差为6.76 eV,HOMO-LUMO能级间电子跃迁的激发线波长λ=293.92 nm.分子静电势分析结果表明哈尔满碱分子有9个极大值点,极大值点是24号H原子,极大值能量为51.322 kcal/mol,有7个极小值点,极小值点的位置是18号N原子,极小值能量为-38.804 kcal/mol.采用TDDFT方法计算了该分子在不同溶剂条件下的紫外吸收光谱和激发态.采用含时密度泛函理论对哈尔满碱分子进行了激发态分析计算,结果表明随着溶剂极性的降低,吸收波长发生红移.为研究哈尔满碱分子的电子结构、光谱性质和分子识别提供了理论依据.
