双位点配位聚合物改性的无钴富锂锰基正极材料 电化学性能研究
2022-03-03张思玉陈敏健马骋张春晓韦伟峰
张思玉,陈敏健,马骋,张春晓,韦伟峰
(中南大学 粉末冶金国家重点实验室,长沙 410083)
近些年来,锂离子二次电池在便携式电子产品、电动汽车等技术领域得到了快速发展,但是由于人们对高能量密度锂离子电池的要求,高比能正极材料和电解质的界面问题成为当前的研究热点[1−2]。尽管早在1991年LiCoO2正极材料已经被商业化应用,但是其可逆容量低(在充放电到4.2 V vs Li/Li+时,容量仅为 140 mAh/g),而且钴价格昂贵[3−4]。因此,研究者们把目光放在了LiCoO2材料的替代物上。在众多层状正极材料中,富锂锰基层状正极材料xLi2MnO3∙(1−x)LiMO2(M=Mn, Ni, Co等,LLOs)凭借其较高的可逆比容量、较高的工作电压以及室温下储存性能优异而受到广泛关注,被视为最有前景的正极材料之一[5−6]。
LLOs的超高比容量不仅是由阳离子的氧化还原提供,而且晶格氧的氧化还原也提供了额外的电荷补偿[7−10]。但目前LLOs大规模商业化应用还受限于以下几个瓶颈问题:1) 在循环过程中Li2MnO3层状相会逐渐向尖晶石相和岩盐相转变,导致材料出现严重的电压衰减[11];2) 由于Li2MnO3相在首次活化过程中存在不可避免的Li 损失,因此首次库伦效率(Initial Coulombic Efficiency,ICE)只有75%左右[12−13];3) 较差的电子/离子电导性、严重的界面副反应以及相变导致了材料的循环稳定性和倍率性能较差[14−15];4) 富锂材料颗粒在高温下循环时界面副反应加剧,O2释放严重;在低温下Li2MnO3相活化受阻,材料的充放电容量很低,其温度适应性较差[16−17]。
很多研究者针对上述问题开展了改性研究,包括表面包覆、元素掺杂、纳米化及构造复合结构等手段,在这些改性手段里面,表面包覆被证明是一种行之有效的方法。目前正极材料的表面包覆主要包括无机包覆和有机包覆,无机包覆包括碳材料[18−19]、金属氧化物[20]、金属磷酸盐[21]、金属氟化物[22]、金属氧氟化物[23]、金属氢氧化物[24]等。Richard团队[25]借助原子层沉积法(Atomic layer deposition, ALD)在Li1.2Ni0.13- Mn0.54Co0.13O2粉末上分别涂覆Al2O3和TiO2,膜层均匀致密,但成本较高。与无机包覆相比,有机包覆更加均匀、操作简单、经济。ZHAO等[26]通过使用带有强电子基团的1,3,6-己三腈(1,3,6- hexanetricar- bonitrile,HTCN)作为电解质添加剂,借助HTCN的三个氰基和LLOs中的TM离子的配位作用对其进行锚定,同时在三(三甲基硅烷)磷酸酯(tris(trimethylsilyl) phosphate, TMSP)的作用下重新构建CEI层,避免正极受各种界面副反应的影响。因此如果能够借助这种具有配位作用的有机物对正极材料进行包覆,引入强配位基团与TM离子络合,在降低TM离子催化活性的同时减少其进一步溶解,实现对TM离子溶解−沉积的双重抑制作用,提高正极/电解质的界面稳定性,使正极材料的循环稳定性大幅提升。
然而由于当前大部分聚合物电解质与高电压层状正极材料表面接触时会发生严重的氧化还原分解,有机物包覆层状正极材料之间的界面研究还比较有限[27]。本文提出一种在富锂锰基正极材料上原位聚合双位点配位聚合物的改性策略:采用溶液原位聚合形成高抗氧化性的聚乙酰乙酸甲基丙烯酸乙二醇酯(PAAEM)聚合物实现对富锂锰基层状材料的表面包覆,通过强配位基团对正极材料表面的TM离子进行锚定,进而形成一层紧密接触的有机物包覆层。通过调节有机螯合材料的加入量优化材料制备工艺,改善富锂锰基层状材料表面的界面稳定性,减少TM离子的溶出,提高材料电化学性能。
1 实验
1.1 材料准备
原始LMNO(Li1.2Mn0.53Ni0.27O2)样品合成:采用共沉淀法[28]制备碳酸盐前驱体(Mn0.53Ni0.27)CO3,将化学计量比为2:1的MnSO4·H2O、NiSO4·6H2O溶解在一定量的去离子水中,得到浓度为2 mol/L的过渡金属溶液,同时配制2 mol/L的碳酸钠溶液、0.24 mol/L的氨水溶液,3种溶液同时泵入反应釜中,控制温度为60 ℃,pH为7.8±0.5。反应10 h后,将沉淀物洗涤干燥,得到碳酸盐前驱体。再将合成的前驱体和Li2CO3混合,在500 ℃煅烧5 h,再在880 ℃煅烧12 h,冷却到室温得到原始LMNO。
PAAEM包覆LMNO步骤:将一定量的有机螯合单体乙酰乙酸甲基丙烯酸乙二醇酯(AAEM,1%,2%,3%,质量分数)和1 g LMNO放在三口烧瓶中,随后加入20 mL的无水乙腈,在氩气保护下搅拌混合均匀,再加入一定量的引发剂,于60 ℃加热4 h使反应完全。反应结束后,用旋转蒸发仪加热到60 ℃除去无水乙腈溶剂得到固体粉末,于70 ℃真空干燥12 h,得到不同质量分数(1%、2%、3%)PAAEM包覆的LMNO(LMNO @PAAEM)。
1.2 电池组装和电化学性能测试
将正极活性材料、聚偏氟乙烯(polyvinylidene fluoride, PVDF)、导电炭黑根据8:1:1的质量比混合均匀溶解在一定体积的N-甲基吡咯烷酮(N-Methyl- 2-Pyrrolidone,NMP)中,充分混合后[29],均匀涂布于干净的铝箔上,干燥后裁剪成直径为12 mm的圆片,辊压后在110 ℃真空烘箱内干燥12 h,得到极片。在氩气气氛的手套箱内组装CR2016扣式电池。采用1 mol/L LiPF6溶于EC/DMC(体积比为1:1)的混合溶液为电解液。电池在30 ℃进行电化学测试,电化学测试在Land系统上进行,在2~4.65 V(vs Li+/Li)以0.5C进行充放电测试。电化学阻抗谱(electrochemical impedance spectroscopy, EIS)使用PARSTAT 4000电化学系统(普林斯顿应用研究公司)进行,频率为0.1~100 000 Hz,室温下交流振幅为5 mV。
1.3 材料分析与表征
采用扫描电子显微镜(scanning electron microscope, SEM)、透射电子显微镜(transmission electron microscope, TEM)表征LMNO正极材料包覆前后材料的形貌,结合能谱分析仪(energy dispersive spectrometer, EDS)对各元素的种类和分布进行分析。用透射电子显微镜、X射线衍射仪(X-ray diffraction, XRD)对材料的物相组成和晶体结构进行表征。X 射线衍射仪测试条件:以CuKα射线为辐射源,步长 0.02 s,停留时间1.2 s,扫描角度范围 10°~80°。并利用Jade6.0软件对 XRD 结果进行处理。同时利用X射线光电子能谱(X-ray photoelectron spectroscopy,XPS)对材料表面的离子价态和含量进行表征。
2 结果与讨论
2.1 形貌与结构表征
图1(a)~(d)所示为Li1.2Mn0.53Ni0.27O2包覆前后材料的表面形貌,未经包覆的原始LMNO球形颗粒表面呈现多孔状态;用PAAEM修饰的LMNO (LMNO @PAAEM)表面的孔洞减少,存在一层连续的有机包覆层。EDS检测如图1(e)~(h),显示LMNO@PAAEM 材料中Mn、Ni、O、C元素分布较均匀,其中C元素均匀分布说明PAAEM在材料表面形成了一层均匀的有机包覆层,其能有效地减少正极材料和电解质的直接接触。

图1 (a), (b) LMNO原始样的SEM图,(c), (d) LMNO@PAAEM改性样的SEM图和(e), (f), (g), (h) EDS图 Fig.1 SEM images of the LMNO-pristine (a), (b), SEM images of the LMNO@PAAEM (c), (d) and EDS of the LMNO@PAAEM (e), (f), (g), (h)
采用TEM对PAAEM聚合物进行表征,图2显示包覆改性后材料LMNO @PAAEM表面有一层厚度约15 nm的均匀无定形连续层,证明在LMNO表面得到 了预期的PAAEM聚合物包覆层,较薄的涂层厚度保证了材料表面良好的电子离子导通。

图2 (a) LMNO-pristine和(b) LMNO@PAAEM的TEM图 Fig.2 TEM images of LMNO-pristine samples (a) and LMNO@PAAEM (b)
图3所示为聚合物单体材料的FTIR图谱,可以看出,其中化学位移在1 129、1 726、3 003 cm−1处对应有机单机材料乙酰乙酸基甲基丙烯酸乙酯的C—O—C、C=O、不饱和C—H基团的特征吸收峰。强电负性基团C=O的引入能够锚定正极材料表面的TM离子,与TM离子形成配位键,这种配位结构可以实现有机包覆层在循环过程中对LMNO材料稳定黏附。
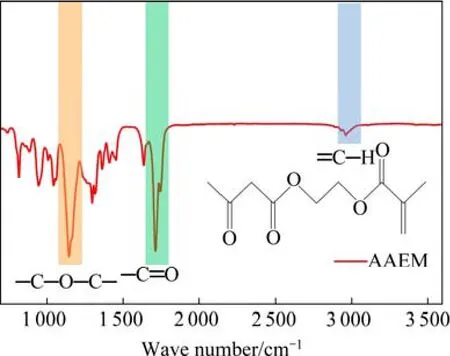
图3 有机单体的红外光谱图 Fig.3 FTIR spectra of AAEM monomer
图4所示为LMNO和LMNO@PAAEM材料的XRD图谱。由图可知,两种材料的XRD图谱类似,和属于R3m空间群的层状α-NaFeO2结构相同,(003)和(104)等特征峰清晰明显,说明其具有较好的层状结构,同时LMNO@PAAEM的 XRD谱图中由于聚合物PAAEM为非晶态而没有出现新的衍射峰。说明PAAEM膜层、PAAEM的合成过程没有改变LMNO 材料的晶体结构。
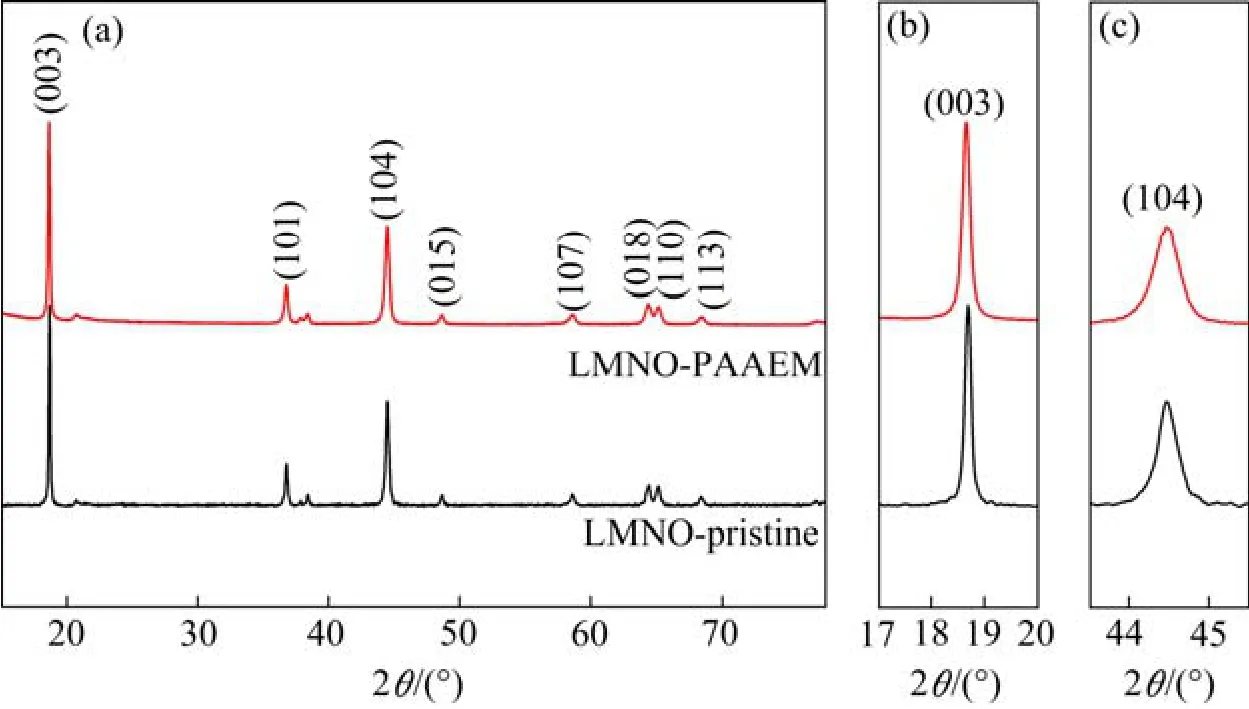
图4 LMNO-pristine和LMNO@PAAEM材料的(a)XRD图及其(b)(003)局部放大图和(c) (104)局部放大图 Fig.4 XRD patterns of LMNO-pristine and LMNO@PAAEM samples (a), enlarged regions for the (003) (b) and (104) peaks (c) of the XRD patterns
图5所示为包覆前后材料表面的XPS图谱,由图可知,采用PAAEM包覆后的材料O、Ni、Mn峰都发生了一定程度的变化。图5(a)~(d)所示的O 1s峰变化明显,531.1 eV和529.6 eV分别代表LMNO材料内部的晶格氧和材料表面的氧空位,532.3 eV和533.8 eV分别代表有机物PAAEM的强电负性基团—C=O和—O—C=O。经过包覆改性后,晶格氧和材料表面的氧空位这两个氧特征峰的强度都有一定程度的减弱,同时PAAEM的两个特征峰—C=O和—O—C出现,进一步证实了聚合物PAAEM在LMNO材料表面的有效包覆。图5(e)~(g)是Mn 2p的XPS图谱,由图可知,LMNO原始材料的Mn4+和Mn3+的特征峰的结合能为642.7 eV和641.5 eV,而包覆改性材料的Mn4+和Mn3+特征峰的结合能为642.9 eV和641.7 eV,向高结合能偏移了0.2 eV左右,说明聚合物包覆层的配位基团和LMNO表面的Mn产生了配位络合作用,使LMNO材料表面的化学环境发生改变,这是因为PAAEM的β-酮酸酯有很强的给电子能力,可与LMNO材料表面的锰离子配位形成稳定的六元螯合环多元配位结构,稳定锰离子。同样图5(h)~(j)中Ni的化学状态也发生了变化,经过包覆改性后,Ni3+和Ni2+的特征峰从855.4 eV和854.3 eV偏移到了更高结合能855.5 eV和854.5 eV处。说明PAAEM的配位基团对LMNO的Ni同样产生了配位作用。从图6可见,改性材料中Mn、Ni峰强进一步减弱后,Mn3+和Ni3+的相对量比例也进一步增加,其中Mn3+的相对量比例增加更多,说明在LMNO材料中,配位聚合物包覆后配位基团对Mn的配位作用较强。
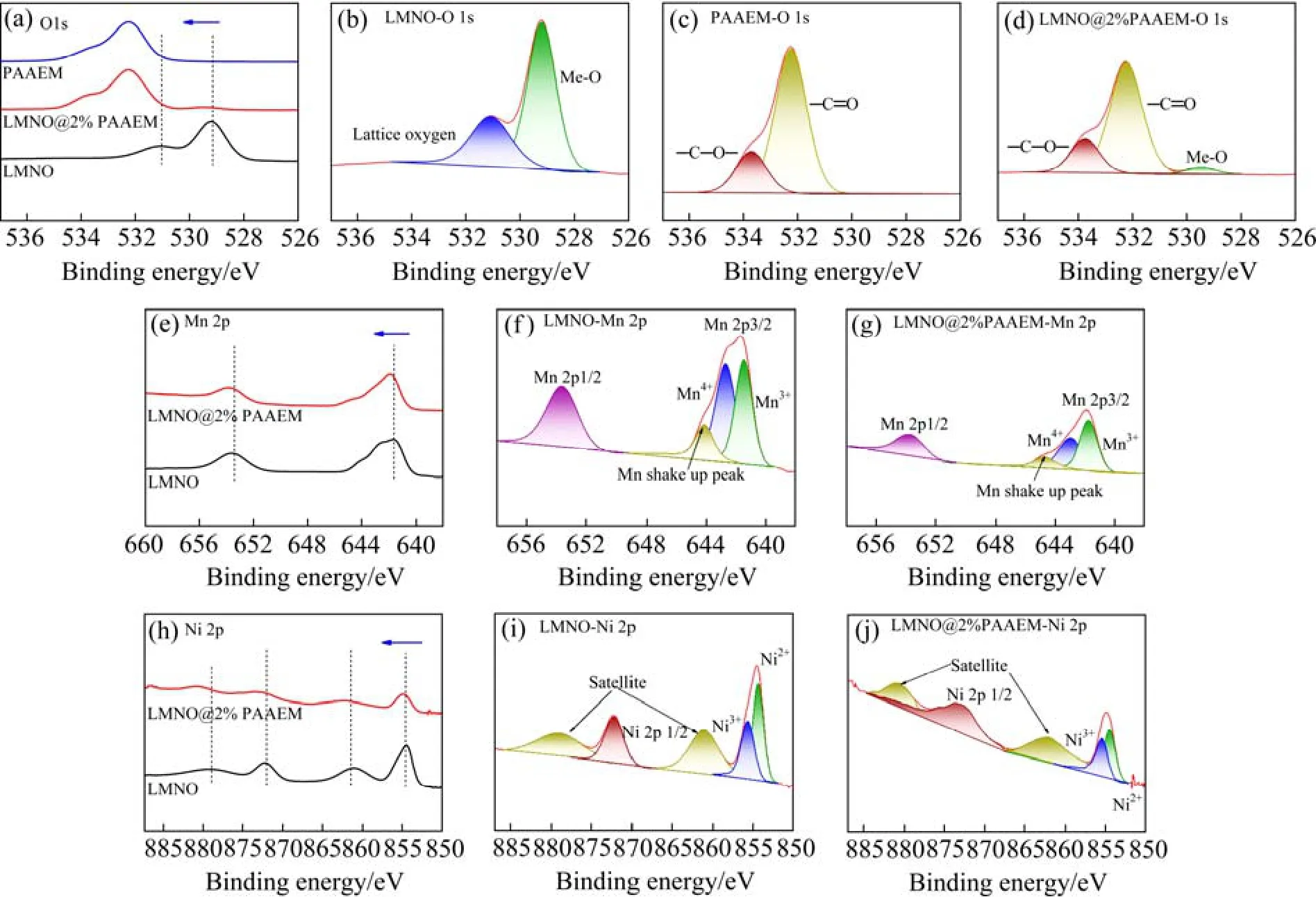
图5 LMNO、PAAEM和LMNO@2%PAAEM的XPS图谱 Fig.5 XPS spectra of LMNO, PAAEM and LMNO@2%PAAEM
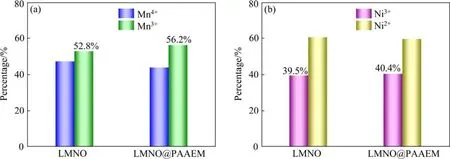
图6 (a) Mn4+/Mn3+化学计量比;(b) Ni3+/Ni2+化学计量比 Fig.6 Stoichiometric ratios of Mn4+/Mn3+ (a) and Ni3+/Ni2+ (b)
2.2 电化学性能测试分析
为了研究PAAEM不同包覆量对LMNO材料电化学性能的影响,在30 ℃下采用恒流充放电测试对其进行电化学性能评估,图7(a)所示为LMNO及不同聚合物比例的LMNO@PAAEM材料在0.5C倍率下的循环性能。结合图7和表1可以看出,LMNO@2%PAAEM材料的循环稳定性最佳,在300圈循环后,原始材料的容量衰减至138.1 mAh/g,容量保持率仅为67%,而LMNO@2%PAAEM容量仍有146.7 mAh/g,容量保持率为82%。这是因为PAAEM的强电负性基团β-酮酸酯能够锚定高活性的TM阳离子,抑制TM离子的进一步溶解析出,减少了电极/电解质的界面副反应,从而形成稳定的CEI界面,提高材料的循环稳定性;另外,聚合物包覆层能够作为物理包覆层有效降 低电极和电解液的直接接触,但随包覆量的增加,LMNO材料表面的有机非晶包覆层厚度增加,离子电导率更低,导电性下降,容量损失更多。图7(b)为LMNO及包覆不同质量分数的PAAEM后的材料在0.5C倍率下的放电中位电压变化情况,从图中可以看出,原始材料LMNO的放电中位电压经历300圈循环后,由3.719 V衰减至3.125 V,电压保持率为84%,而LMNO@2%PAAEM的电压保持率提升至92%。可见PAAEM的包覆对LMNO电压衰减的抑制效果 明显。
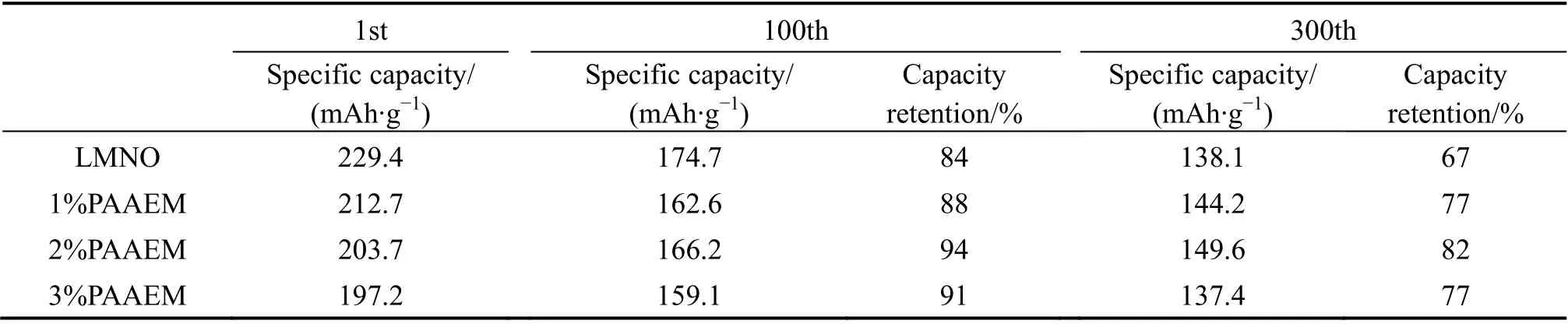
表1 不同包覆量的LMNO材料在100、300圈的容量保持率情况 Table 1 Cycle performance of different percentage PAAEM coated LMNO materials
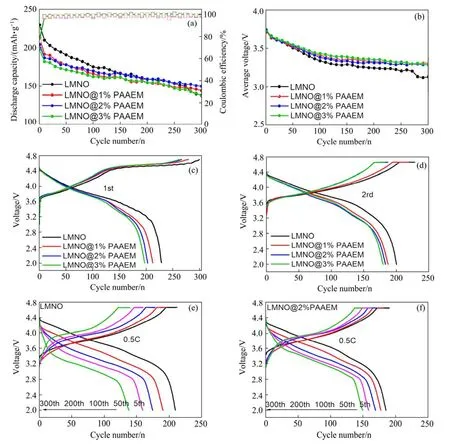
图7 LMNO原始材料和LMNO@PAAEM材料的电化学性能 Fig.7 Electrochemical performance of LMNO-pristine sample and LMNO@PAAEM materials
图7(c)为LMNO材料包覆前后在0.1C、2~4.7 V电压区间的首圈充放电曲线,在4.5 V以下有一个倾斜区域,这是Ni2+氧化为Ni4+的过程,在4.5 V以上存在一个较长的充电平台,这个电荷平台在接下来的循环中消失。一般认为,这个平台是Li2MnO3相的电化学活化,该过程中Li+的脱出和氧气的释放是不可逆的。随PAAEM包覆比例增加,该充电平台缩短,这是由于强配位基团对Mn4+的络合作用,—O—C=O能够锚定LMNO材料表面的TM离子,减少Mn氧化反应的同时可抑制氧气的释出,从而使Li2MnO3的活化过程被抑制。图7(e)、7(f)是原始材料LMNO和LMNO@2%PAAEM的第2、50、100、200、300圈充放电曲线,可以看出,原始材料的放电电压和容量衰减比较严重,LMNO@2%PAAEM的电压衰减和容量衰减得到了有效抑制。综上所述,PAAEM膜层对LMNO材料的电化学性能的提升可以归结为两方面:一是有机聚合物上的β-酮酸酯基羰基对正极材料表面的TM离子具有螯合作用,能够有效抑制TM离子迁移与溶解,从而稳定晶体结构;二是PAAEM有机包覆层可以有效地减少正极材料和电解液的直接接触,从而极大抑制电极-电解液界面副反应,提高界面稳 定性。
图8所示为不同充放电曲线得出的容量电压微分曲线(dQ/dV)。图8(a)是改性前后不同包覆比例材料的首圈dQ/dV曲线,可以看出,包覆改性后材料在4.5 V处的氧化峰比原材料在该位置的峰弱,说明Li2MnO3的活化受到抑制,即氧气的释放得到一定程度的抑制。图8(b)~8(d)所示为Ni2+/Ni4+的氧化还原峰,与原始材料LMNO相比(电位偏移270 mV),LMNO@2% PAAEM在Ni2+/Ni4+的氧化峰的电位偏移仅为140 mV,说明循环过程中LMNO表面结构退化严重,从而导致电压衰减,循环性能下降,这进一步证明PAAEM的包覆能够有效减轻界面退化,并降低循环过程中的过电位,提高材料的循环稳定性。

图8 LMNO原始材料和LMNO@PAAEM材料的(a)首圈dQ/dV曲线,(b)第二圈dQ/dV曲线; (c) LMNO原始材料和(d) LMNO@2%PAAEM在不同圈数时的dQ/dV曲线 Fig.8 The dQ/dV curves of the first (a) and the second circle (b), dQ/dV curves of the as-prepared material at 0.1C LMNO-pristine (c) and LMNO@2%PAAEM (d) at different electrochemical cycles
2.3 LMNO正极/电解质界面分析
图9 所示为循环前LMNO和LMNO@2%PAAEM的电化学阻抗谱图。其中Rs代表在高频时的电解质本体电阻,R1代表电极和电解液界面处的电荷转移电阻,W1代表低频时锂离子在电极界面扩散引起的阻抗。从电化学阻抗谱中可以看出,循环前包覆前后材料的电解质本体电阻相近,原始LMNO材料的电解质本体电 阻为0.6 Ω,电荷转移电阻为75.58 Ω。而包覆改性后材料的溶液电阻是2.80 Ω,电荷转移电阻是109.80 Ω,这是因为有机配位聚合物包覆层可降低正极材料表面的离子电导。经过首圈0.1C倍率活化后,原始LMNO材料的界面转移电阻增大至172.99 Ω,而PAAEM包覆改性后的LMNO材料界面转移电阻仅为119.73 Ω。说明有机包覆层可有效减少正极和电解液之间的接触,抑制正极和电解质之间的界面副反应。
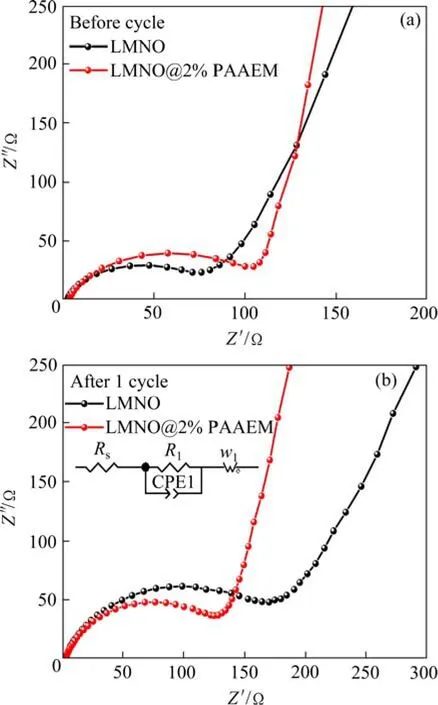
图9 LMNO和LMNO@2%PAAEM循环前后的 电化学阻抗谱 Fig.9 Electrochemical impedance spectra of LMNO and LMNO@2%PAAEM before cycling and after cycling at 0.1C
3 结论
1) 采用溶液原位聚合法在LMNO材料表面构筑了一种具有双齿配位螯合作用的聚合物PAAEM层,聚合物包覆层的厚度在15 nm左右。
2) 聚合物PAAEM的强电负性基团—C=O通过配位络合正极材料表面的TM离子以减少TM离子迁移与溶解,同时锚定溶解的TM离子,与之形成多元螯合环结构,降低TM离子催化活性,减少正极/电解质的界面副反应,保持材料的结构稳定。
3) 与没有包覆改性的材料相比,经过表面聚合物包覆改性的LMNO材料以0.5C循环300圈后容量保持率由67%提升至82%,电压保持率由84%提升至92%,实现了高电压层状正极材料循环稳定性的显著提升。
