以运动后反复血尿起病的极长链酰基辅酶A脱氢酶缺乏症1例报告
2022-03-02向秋莲
丁 乐,郑 帼,梁 超,向秋莲
南京医科大学附属儿童医院神经内科,江苏 南京 210008
极长链酰基辅酶A 脱氢酶缺乏症(very long chain acyl⁃CoA dehydrogenase deficiency,VLCADD)(OMIM#201475)是一种罕见的常染色体隐性遗传病。因发病率低,临床缺乏特异性,往往易漏诊误诊。本研究报道2019年10月南京医科大学附属儿童医院收治的1例运动后反复血尿起病的VLCADD临床资料和基因检测结果,总结该病临床特点并文献复习已报道的病例资料,以提高临床医师对该病的认识,为早期诊断和治疗、遗传咨询提供依据。
1 病例资料
患者,男,11 岁2 月。因“全身肌肉痛伴浓茶色小便3 d”于2019 年10 月17 日入院。3 d 前跑步400 m、踢足球20 min后出现双下肢肌肉疼痛,逐渐蔓延至全身肌肉,伴小便成浓茶色,无尿急、尿频、尿痛,无排尿中断,至南京医科大学附属儿童医院门诊查尿微量蛋白3项、丙氨酸氨基转移酶(ALT)、天门冬氨酸氨基转移酶(AST)、磷酸肌酸激酶(CK)、肌酸酶同功酶(CKMB)等明显升高,门诊予“阿拓莫兰、复方甘草酸苷”保肝治疗,“碳酸氢钠”碱化尿液治疗1 d肌肉酸痛好转,拟诊“横纹肌溶解症”进一步收住本科。病程中无发热,无咳嗽,无呕吐腹泻,食纳可,精神睡眠可,大便未见异常。患儿既往2019 年8 月因“运动后肉眼血尿”收住本科诊断“横纹肌溶解症”,给予对症支持治疗后好转出院。反复追问病史生后至今有运动后4、5次全身肌肉酸痛,休息后好转,未行相关检查。个人史及家族史无特殊,目前有一3岁弟弟,身体健康。入院体格检查:身高162 cm,体重34 kg,神志清,精神一般,营养中等,发育正常,心肺查体正常,腹部查体肝脾无肿大,双侧肾区叩击痛。神经系统检查无异常。实验室检查:血常规、肾功能、降钙素原、凝血常规、血气分析、血糖均正常;泌尿系B超、心脏彩超均未见异常;尿微量蛋白3项:尿微量白蛋白585.00 mg/L,尿IgG 测定78.10 mg/L,尿α1 微球蛋白定量349 mg/L;尿沉渣:深棕色,尿隐血+++,尿蛋白++,尿比重1.045,白细胞计数25.3/μL;血生化:ALT 242 U/L,AST 1 358 U/L,CK 64 400 U/L,CKMB 1 673 U/L,肌红蛋白2 255.7 ng/mL,余未见明显异常。
入院后予注射用还原型谷胱甘肽1.2 g、复方甘草酸苷注射液40 mg 保护肝功能、注射用磷酸肌酸钠1 g营养心肌、碳酸氢钠碱化尿液、果糖二磷酸钠、辅酶Q1O、维生素C 等改善能量对症支持治疗。入院第2天血尿基本消失,全身酸痛很快缓解。1周后复查血生化:ALT 244 U/L,AST 138 U/L,CK 2129 U/L,CKMB 49 U/L,肌红蛋白25 ng/mL。鉴于患儿反复运动后出现横纹肌溶解,其运动强度一般,排除中毒、感染、外伤等诱因,故高度怀疑遗传代谢病可能。
经患儿父母知情同意,医院医学伦理委员会批准后分别抽取患儿、其父母及弟弟2 mL 外周血,送至北京康旭医学检验所进行基因检测,发现患者ACADVL 基因(图1)存在复合杂合突变:1 个c.1146+6T>A剪切突变和1个c.549C>G(p.Tyr183Ter)无义突变。Sanger测序证实,c.1146+6T>A 源于其母亲,c.549C>G 源于其父亲。其弟弟携带一个c.549C>G 杂合突变。随访建议患儿低脂高碳水化合物饮食,避免剧烈运动、感染、劳累、饥饿等,1个月后复查肝功能、肌酶均正常。3个月后有次踢球后肌肉酸痛,自行休息后好转。
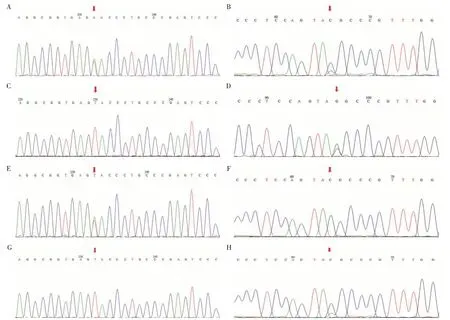
图1 ACADVL基因分析结果
2 讨论
极长链酰基辅酶A脱氢酶(very long chain acyl⁃CoA dehydrogenase,VLCAD)是线粒体脂肪酸β氧化中第一步的关键酶,催化14~20 个碳的脂酰基辅酶A 脱氢,产生的氢原子进入线粒体呼吸链进行氧化磷酸化产生ATP 提供能量。故VLCADD 发病机制为脂肪酸氧化缺陷和随后的酮体合成损伤而造成的严重能量缺乏以及有毒长链酰基肉碱蓄积在细胞内对心肌、骨骼肌、肝脏等产生毒性作用。
VLCADD于1993年由Aoyama等[1]首次发现,其临床表型有明显异质性,可表现为不同的严重程度和发病年龄,从无症状个体到致命结局,可发生在新生儿期至成年期的任一时期。可因长时间或剧烈运动、禁食、发热或疾病而诱发,因此早期诊断,治疗和监测预防可以减少死亡。根据涉及主要依靠脂肪酸氧化产生能量的器官参与程度和起病年龄分为3型[2]:①心肌病型(VLCAD⁃C):多在新生儿或婴儿早期发病,起病凶险,病死率高,表现为低酮症性低血糖、脑病、新生儿猝死、心肌酶升高、肥厚性心肌病、心律失常等;②肝病型(VLCAD⁃H):婴儿晚期或幼儿期起病,以低酮症性低血糖为主,可伴有肝功能异常,症状较轻,少有心脏累及;③肌病型(VLCAD⁃M):青少年或成人期起病,表现为运动不耐受、肌痛、横纹肌溶解、肌红蛋白尿等。本研究中患儿有运动不耐受,反复运动后肌痛以及2 次运动后横纹肌溶解病史,引起我们的重视,完善基因检测后明确诊断为肌病型VLCADD。此型起病于青春期或成年期,症状虽然轻微,但过度运动、应激、饥饿、疾病时会诱发,反复发作势必导致肾损害。
VLCADD 是威胁生命但可治疗的代谢性疾病,因此该疾病已在美国被列为新生儿推荐统一筛查项目中。新生儿筛查(NBS)血斑十四烷酰肉碱水平(C14∶1)明显升高是唯一用于VLCADD 的生化指标[3]。Suzan 等[4]最新研究认为NBS 检测出VLCADD 的新生儿可能具有严重的表型,也可能不会出现症状。他们通过代谢组学早期预测其严重表型,确定了C18∶2 肉碱和C20∶0 肉碱作为预测疾病严重程度的生化标志物。Pena等[5]回顾性分析了由NBS诊断为VLCADD的52例年龄在1~18岁之间患者最初的表现、诊断、临床结果和治疗。在该队列中,2/52例被诊断心肌病,其中1例于11.5个月死于急性呼吸衰竭。同时CK升高是此类患者一个常见的表现,通常首次发生在幼儿期(1~3岁)。在14例CK 升高的受试者中,有11 例出现了横纹肌溶解症(CK≥1 000 μmol/L)。故临床中应该关注不能被其他疾病所解释的CK升高的婴幼儿。
VLCADD 诊断评估方法最常见的是血浆酰基肉碱谱和ACADVL 基因分子检测。目前随着全外显子测序技术发展,基因突变分析是确诊VLCADD的金标准。VLCADD由ACADVL基因(OMIM609575)编码。ACADVL 基因位于染色体17p13.1,长约5.4 kb,含20个外显子,编码655个氨基酸前体蛋白,位于线粒体内膜属于脂酰辅酶A脱氢酶(acyl coen⁃zyme A dehydragenase,ACAD)家族成员之一。到目前为止,已报道ACADVL 基因321 种致病和可疑致病突变(HGMD,http://www.Hgmd.cf.ac.uk/ac/in⁃dex.php,2019年6月),其中以错义变异为主。本例患者ACADVL基因存在复合杂合突变,1个c.1146+6T>A 剪切突变和1个c.549C>G(p.Tyr183Ter)无义突变。c.1146+6T>A 虽然在内含子11 区,但其致病性已有文献报道[6]。另1个c.549C>G(p.Tyr183Ter)为无义突变,导致蛋白翻译提前终止,虽未有文献报道,但结合患儿临床表型及常染色体隐性遗传方式,从生物学意义上推测p.Tyr183Ter 是致病性突变,且是一个新发突变。
VLCADD 的治疗原则是避免空腹、感染、脱水、高脂饮食和过度运动疲劳,给予高碳水化合物、低脂补充中链三酰甘油(medium⁃chain triglycerides,MCT)的饮食,对症处理及预防并发症。高温会使VLCAD 活性降解[7],因此,将发热的VLCADD 患者的体温降低至正常水平至关重要。Watanabe等[8]近期报道了2 例VLCADD 同胞在补充左旋肉碱后发生横纹肌溶解。目前尚不清楚在VLCADD 患者中使用肉碱是否安全以及预防肉碱缺乏的益处是否超过长链酰基肉碱累积的潜在心脏风险。虽然补充肉毒碱被认为存在争议,但实际大多数患者都在接受肉碱补充。Li等[6]研究中表明当患者血肉碱水平<20 mmol/L时,可以服用L⁃肉碱(每天20~50 mg/kg),为避免L⁃肉碱的不良反应,需仔细随访生化和临床情况。过氧化物酶体增殖物激活受体的激动剂苯扎贝特可增加VLCAD蛋白和mRNA表达[9]。大多数患者都服用苯扎贝特,并配以富含MCT的配方L⁃肉碱,葡萄糖醛酸内酯,维生素C和维生素B复合物。此外,定期对心脏、肌肉和肝功能的严格监控将确保良好的预后,因为器官功能障碍通常可以通过治疗逆转。
