Rational design of heterogeneous catalysts by breaking and rebuilding scaling relations
2022-03-01WeiQiYanYiAnZhuXingGuiZhouWeiKangYuan
Wei-Qi Yan,Yi-An Zhu,Xing-Gui Zhou,Wei-Kang Yuan
State Key Laboratory of Chemical Engineering,School of Chemical Engineering,East China University of Science and Technology,Shanghai 200237,China
Keywords:Density functional theory (DFT)Heterogeneous catalysis Microkinetic analysis Scaling relations
ABSTRACT Various scaling relations have long been established in the field of heterogeneous catalysis,but the resultant volcano curves inherently limit the catalytic performance of catalyst candidates.On the other hand,it is still very challenging to develop universal descriptors that can be used in various types of catalysts and reaction systems.For these reasons,several strategies have recently been proposed to break and rebuild scaling relations to go beyond the top of volcanoes.In this review,some previously proposed descriptors have been briefly introduced.Then,the strategies for breaking known and establishing new and more generalized scaling relations in complex catalytic systems have been summarized.Finally,the application of machine-learning techniques in identifying universal descriptors for future computational design and high-throughput screening of heterogeneous catalysts has been discussed.
1.Introduction
Heterogeneous catalysts have been playing an important role in the chemical industry,which may have the capability of increasing the rate of chemical reactions and improving the selectivity toward desired products[1,2].On the other hand,in various catalytic reactions,noble metals such as Pt and Pd could be active and selective,but their high cost limits their practical application.Hence,researchers have been paying more attention to the discovery of non-precious metal catalysts for a specific catalytic reaction.In the past,the development and optimization of catalysts depends strongly on chemical intuition and experience which is based on a trial-and-error method.For this reason,a long-standing goal of heterogeneous catalysis is to design catalysts for a given reaction in a more rational and efficient way.
It is well known that many factors such as catalyst compositions and structures,support properties,and reaction conditions may affect the catalytic performance of the catalysts,which makes it difficult to predict the trend in the catalyst activity and selectivity based on the understanding of the reaction kinetics.Nowadays,computational modeling provides a way to discover new and more effective catalysts.By combining quantum chemical calculations[mostly based on density functional theory (DFT)] [3] with microkinetic analysis or kinetic Monte Carlo simulations,detailed reaction mechanisms can be determined at the molecular level.In such kind of calculations,although the energetics of the elementary steps in the catalytic network can be used to calculate with reasonable accuracy the activity of the catalyst,it is too timeconsuming to calculate the activity of each catalyst individually and apply this method in high-throughput screening of catalyst candidates and,more importantly,one would get lost in the detailed kinetic data and cannot capture the key to the success of the catalyst screening.For this reason,a key step to screen potential catalysts is to identify reactivity descriptors by establishing scaling relations.Electronic and geometrical properties of catalysts and adsorption energies of simple species may sever as the descriptor(s),which can then be used to describe the energetics of elementary steps involved in the reaction network and thus the kinetics of the overall reaction.In this way,the higherdimensional “parameter space” can be mapped into a lowerdimensional “descriptor space”,and the reaction kinetics has its complexity hidden in the resultant volcano-shaped curves(or contours) as a function of one or two descriptors.
So far there have been many successful examples using volcano-shaped plots as a tool to screen new catalysts [4,5],but almost all of these studies have been conducted over a single family of compounds,such as transition metals and alloys [3,6–8],transition-metal oxides [9–11],and carbon-based materials [12–14],etc.The reason lies in the fact that scaling relations established can only hold on materials of similar structures and properties.It is still very challenging to develop universal descriptors that can be used in various types of reaction systems over various catalysts.On the other hand,scaling relations inherently limit the catalytic performance of catalyst candidates,making it difficult to achieve simultaneously low activation energy barrier and weak intermediate adsorption strength,so that the resulting volcano-shaped plots cannot predict catalysts that show stronger activity than the “optimal” catalyst.From a mathematical perspective,Mao et al.analyzed the origin of the volcano curve by using a two-step kinetic model,and proved that the volcano curve is caused by the rapid occupation of the active sites with increasing adsorption strength[15].Hence,only when scaling relations are no longer valid under some circumstances,it is possible to go beyond the summit of volcanoes.Hence,several attempts have recently been made to break scaling relations,but it is very likely that new scaling relations can be establishing at a different(higher)level,which in turn gives rise to a new volcano curve and a better catalyst candidate.
In this review,the previously proposed descriptors in the field of heterogeneous catalysis have first been summarized as shown in Fig.1.Then,special attention has been devoted to the strategies proposed to break scaling relations and establish new scaling relations.Finally,the possible application of the machine-learning method in identifying universal descriptors for future computational design and high-throughput screening of catalysts has been discussed.
2.Previously Proposed Descriptors
In the 1990s,an electronic descriptor based on the dband model for transition metal catalysts was first proposed by Hammer and Nørskov [19–23],which connects the binding strength of adsorbates with the d-band electronic structure of transition metals.More specifically,both the strength of the adsorbate–metal interaction and the dissociation energy barrier are related to the degree of filling of the antibonding states upon adsorption and the degree of orbital overlapping with adsorbates [20].Since then,the dband model has been widely used to describe the catalytic activity of transition-metal catalysts.On the basis of this model,the average energy of the dband,namely,the dband center,was proposed and used as a reactivity descriptor on transition-metal catalysts[3,24].For example,if the outermost layer on Pt(111) is replaced by 3d metals,the d-band is broadened and lowered in energy,resulting in weaker dissociative adsorption energies of hydrogen and oxygen [3].Hence,the d-band centers of modified Pt surfaces scale well with their dissociative adsorption energies.Moreover,because the change in the activation energy for elementary steps was suggested to be proportional to the change in the reaction enthalpy [the Brønsted-Evans-Polanyi (BEP) relation [16,25–29],the energy barriers can also be estimated by calculating the adsorption energies of the species involved in the reaction,which are in turn correlated with the d-band centers.Thus,d-band center is a good reactivity descriptor which can predict trends in the catalytic performance and give theoretical guidelines for catalyst design.In addition to the d-band center,some other descriptors based on the d-band model,such as the width and shape of the d band,have also been found to be of importance in determining the reactivity of transition-metal alloys.
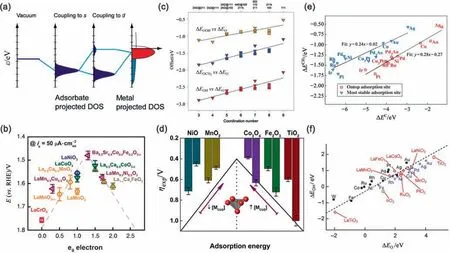
Fig.1.Previously proposed descriptors.(a)A schematic illustration of bond formation on a transition-metal surface.The lower the d states are in energy relative to the Fermi level,the more filled the antibonding states,and hence the weaker the adsorption bond.(b)A volcano plot depicting the relation between the catalytic activities of the oxygen evolution reaction overpotential as a function of the reversible hydrogen electrode (RHE).(c) The offsets of the scaling relations between the adsorption energies of *OH versus *O (red),*OOH versus *O (orange),and *OCH3 versus *O (blue) scale in an approximately linear fashion with the coordination number of the adsorption sites.(d) A volcano plot of the overpotential in the oxygen evolution reaction as a function of the density of coordinatively unsaturated metal cation of transition-metal oxides.(e)Binding energies of CH3 plotted against the binding energies of C for adsorption at the most stable sites(triangles)and in the case where both CH3 and C have been fixed in the on-top site(squares).(f)Adsorption energies of the OH intermediate on transition-metal oxide and transition-metal surfaces are plotted against the adsorption energies of O[2,6,11,16,17,18].(For interpretation of the references to colour in this figure legend,the reader is referred to the web version of this article.)
As for metal oxides,it is more challenging to establish scaling relations because of the higher complexity of the electronic and geometrical properties of metal-oxide surfaces.For instance,perovskite oxides have been widely used in water splitting [30–33],ORR and oxygen evolution reactions (OER) for fuel cells [34–36],and partial oxidation of methane[37].To predict the OER intrinsic activity of perovskite oxides,it has been proposed that the filling of the antibonding states of the egorbitals,called eg-filling,is an excellent electronic descriptor which scales well with the adsorption energies of oxygenated species [17].In addition,as an important bulk property of the material,band gap has been successfully applied to scale with the activation energy for the rate-limiting step in propene oxidation on mixed-metal oxides.Combined with the enthalpy of formation of oxides,the band gap is capable to predict the oxygen vacancy formation energy in LaxSr1-xBO3perovskites (where B=Cr,Mn,Fe,Co,and Ni) [38].
The descriptors mentioned above are determined by electronic properties and are therefore considered as electronic descriptors.Other electronic descriptors include charge polarization [39] and the average 2p-state energy[40].Considering the significant influence of structural properties on the catalytic behaviors of materials,structural descriptors have also been proposed and widely used in structure–sensitive systems [8,41].For example,the coordination number(CNs)of the binding atoms in active sites exhibit linear scaling relations with the adsorption energies of oxygen and oxygenates on transition metals [42].By counting the CNs of the atoms in the active sites,the binding strength towards reaction intermediates can be predicted,which is then transferred to the adsorption–energy scaling relations to predict catalytic reactivity.Due to the complexity of transition-metal oxides surfaces,the CN descriptor should be adjusted to predict the catalytic activity of metal oxides.The adjusted CN involves not only the CN of the surface oxygen site (CNO) but also the neighboring metal atoms(∑CNM) [43].Five facets of four transition-metal oxides (V2O3,Cr2O3,Co3O4and NiO) can be described based on the adjusted CN,and the C-H activation energy on Co3O4is predicted based on this structural descriptor.Another structural descriptor has been identified on transition-metal oxides for OER is the density of coordinatively unsaturated metal cations on oxide surfaces(MCUS).By modifying the MCUSlevel,the adsorption of intermediate can be tuned,thereby achieving the optimal OER activity.
Adsorption energy provides a measure of how strongly species are bonded to the catalyst surface[44].To reduce the dimensionality of “adsorption energy space” and reveal the nature of the adsorbate–substrate bonding,scaling relations between the adsorption energies of CHx(x=0,1,2,3),NHx(x=0,1,2),OHx(x=0,1)and SHx(x=0,1) have been proposed over a wide variety of transition-metal surfaces.The adsorption energies of these hydrogenated molecules correlate well with the adsorption energies of the central atoms (C,N,O,and S) [6].As a result,it is possible to construct the full potential energy diagram for a surfacecatalyzed reaction by using the C,N,O,and S chemisorption energies as descriptors.For example,the adsorption energies of oxygen reflect the stability of the Pt-OOH,Pt-OH,and Pt-O bonds,which can be used to estimate the overall activity of oxygen reduction reaction (ORR) on Pt-based bimetallic catalysts [45].Similarly,the adsorption energies of H have also been used as an activity descriptor to successfully guide the electrocatalyst screening for the hydrogen evolution reaction (HER) [46–48].As for metal oxides,it has been found that the binding energy of *OH and *OOH on rutile-type oxide (TiO2,IrO2and RuO2) in the water-splitting reaction are linearly correlated with the binding energy of*O.Furthermore,the adsorption energies of O,OH,S,SH,N,NH,and NH2on transition metal oxide,sulfide,and nitride surfaces are calculated,and a scaling relation was found between the adsorption energies of the intermediates and the adsorption energies of the atoms,which is independent of the metal and depends only on the number of H atoms in the molecule[11].In addition to adsorption energy,the oxygen vacancy formation energy can be taken as a reactivity descriptor for the partial oxidation of methane.For (K,Rb,Cs,Sr,Ba)BO3(B=d-block transition metals)and ARuO3(A=La to Ho) with various crystal structures,the oxygen-vacancy formation energies scale well with the adsorption energies of C,H,O,and CH3at the O site [49].
3.Breaking of Scaling Relations
As aforementioned,theoretical approaches to rational design of catalysts are based on the identification of a few descriptors to construct the activity and selectivity volcanoes.The binding strength of reaction intermediates to the surface can be readily obtained from scaling relations,while the BEP relationship gives correlation between the activation barrier and reaction heat of elementary steps,making it possible to predict the overall reaction rate.However,the scaling relations established also limit the catalytic performance of catalysts.To obtain catalysts with better catalytic performance than those lying near the summit of the volcano,the scaling relations need to be broken.
The simplest way to break scaling relations is through the geometrical effect.Because of the restriction of scaling relations,the enhanced catalytic activity is often accompanied by the acceleration of side reactions,sometimes leading to a low selectivity toward target products.To obtain excellent catalytic activity and selectivity simultaneously,the structural properties of the catalyst surface can be tailored to the desirable main reactions,particularly in the situations where the side reactions might require larger ensembles than the main reactions [50–52].For instance,alkyne hydrogenation takes place at very small active ensembles,but C-C bond formation in the alkyne molecules needs more than six transition-metal atoms [53].Therefore,limiting the size of ensembles may improve the catalyst selectivity by breaking the positive correlation of activities between alkyne hydrogenation and C-C bond formation.In essence,due to the size effect,a new scaling relation for C-C bond formation activity is established in this catalytic system.Another example of the application of the geometrical effect is single-atom catalysts,which commonly have reactive centers surrounded by inert atoms.As a consequence,the reactants can dissociate on the reactive metal atoms and the target products can easily desorb from the coordination sites on singleatom catalysts.For example,after single platinum atoms are embedded in copper nanoparticles,the desorption of propylene is found to be enhanced while its further dehydrogenation is prohibited,which breaks the scaling relations for PtM (where M=3d and 4d transition metals) alloys and results in higher propylene selectivity than the conventional Pt alloys[54].In addition,scaling relations can be broken on the sub-nanoscale,due to the highly structural fluxional behavior of sub-nanometer-sized clusters.More specifically,sub-nanometer-sized clusters may adapt their structures to the bound adsorbates and their varying coverage,or even reshape the geometry as the environment changes,which in turn breaks the scaling relations between the binding energies of key intermediates.It has been suggested that on the sub-nanometer-sized Ptnclusters the binding energies of the reaction intermediates involved in the ORR reaction,such as O,OH,and OOH,no longer scale with each other[55].This fluxional behavior is very common on clusters,suggesting that breaking scaling relationships is likely a rule rather than an exception in nanocluster catalysis due to the geometrical effect.Alternatively,scaling relations can be broken by modifying the electronic structures of catalyst surfaces.On alloys,altering the surface composition is an effective way to modify the electronic structures and hence to break the inherent scaling relations.For instance,Pd0.75-Ag0.25has obviously weakened *CO and *H bindings but retains well the binding with *COOH among several Pd1-xAgx,which contributes to the improvement in the CO selectivity in the CO2reduction reaction.On transition-metal oxides,Lewis acid-base interactions were often used to break scaling relations.The adsorption and catalytic behaviors of single-atom-doped Ga2O3catalysts in propane dehydrogenation shows that the scaling relations can be broken in the presence of the Lewis acid-base interactions,making it possible to achieve better catalytic performance than the optimal catalyst predicted in the absence of Lewis acid-base interactions[56].In addition,the unique electronic properties of semiconductors can also break the scaling relations resulting from metal-like materials.For example,the Br version of the Deacon reaction on the oxides of the rutile family consists of five steps including surface oxidation,the adsorption of the acid,the halogen evolution,hydroxyl recombination,and water elimination.According to this reaction mechanism,the reaction can hardly occur on the TiO2rutile,a semiconductor that cannot absorb oxygen [55].However,when HBr is adsorbed on the rutile,the system is self-doped,leading to the breaking of the scaling relations between O and Br adsorption energies and hence enhancing the adsorption of oxygen[56].
Another method to break scaling relations is to alter the reaction mechanism by adding a second active component or agents[57].To break the linear scaling relationship of intermediate binding and minimize the kinetic barrier for CO2reduction reactions for electrocatalytic ethylene production,ternary Cu-Au/Ag nanoframes were fabricated to decouple the functions of CO generation and C-C coupling,where the former is promoted by the alloyed Ag/Au substrate and the latter is facilitated by the highly strained and positively charged Cu domains.For OER,the change of mechanism is even more thorough.On both metallic and non-metallic catalysts,there are scaling relations between the overpotential of the full OER and the Gibbs free energy change for reaction steps.On certain combinations of catalysts,the scaling relations have been broken due to the newly discovered bifunctional mechanism formulated,leading to the reduction in the OER overpotential [58].As for layered double hydroxides (LDHs),which are among the most active and studied catalysts for OER in the alkaline electrolytes,the OH-O scaling relationship can also be broken by forming binary metal oxyhydroxides with dual metal sites as the reaction centers,or by introducing a third element into LDHs.Also,one may decorate transition-metal surfaces with agent to break scaling relations.For example,the scaling relations that limit the performance of ammonia synthesis on transition metals can be broken by LiH [59].The negatively charged hydrogen in LiH not only acts as a strong reducing agent in the reaction to remove the activated nitrogen atoms from the metal surface,but also serves as a direct hydrogen source,which makes it possible for ammonia synthesis to occur under mild conditions.Inspired by this work,it was reported that a bifunctional TiO2-xHy/Fe catalyst,in which the Fe component enables facile activation of N2and the OV-H in TiO2-xHyhydrogenates the N to NH3readily,is able to break the scaling relation in ammonia synthesis.
Finally,the use of external forces can help to break scaling relations.Plasmonic metal nanoparticles are found to be able to drive new transformations effectively.The incoming electromagnetic radiation in the form of light concentrates in the form of plasmons and therefore results in new activity rules [60].In addition,the oxygen evolution reaction on antiferromagnets at high pH has been found to be enhanced by magnetic effects due to spincontrol in the reaction.
4.Rebuilding Scaling Relations
When scaling relations are broken,new physical principles are needed so as to design better catalysts.In reality,the breaking of known scaling relations implies the possibility of establishing new scaling relations.After the reason why scaling relations would fail in some catalytic systems has been elucidated,several new substituents have been proposed as shown in Fig.2.
The high degree of site heterogeneity on the catalyst surface explains why scaling relations are broken.For most structural descriptors,they are only valid in some systems of similar structures.The CN is rather a common structural descriptor as mentioned above,but it is not a universally applicable descriptor as well because of the well-known finite-size effects[61,63].To avoid this problem,the concept of generalized CN is proposed [61] to design Pt-based catalysts for ORR,which not only is related to the coordination number of the central atom but also to the coordination number of the nearest neighbor atoms.Also,the orbitalwise CN(CNα,α=s or d)has been developed based on CNs,which quantifies both the coordinative saturation degree of metal atoms and their tendency to form bonds via the α orbital of a specific adsorption site.It can be applied to complex systems with varying lattice strains and metal ligands [18].
On the other hand,it is likely that some important electronic properties of a specific catalytic system cannot be properly described by the known scaling relations.Under such circumstances,searching for accessible properties that are closely related to these electronic properties is a very effective way to identify new descriptors.For example,the expected number of electrons transferred to and from oxygen to perovskites cannot be described by d-band center,which causes the failure to predict the areaspecific resistance on perovskites.As a result,the p-band center of lattice oxygen was proposed instead as an electronic descriptor to establish better linear relationships with experimental surface exchange coefficients [62],which is more readily obtained as a bulk property.The high OER activity of double perovskites (Ln0.5-Ba0.5)CoO3–δ(Ln=Pr,Sm,Gd and Ho) in the alkaline solution was attributed to the appropriate O p-band center,which is neither too near nor too far from the Fermi level [64].In general,O pband center tends to be a better descriptor than d-band center or eg-filling on metal-oxide catalysts,especially when the electronic properties of lattice oxygen are essential to the catalytic performance.For single-atom-doped Ga2O3catalysts in propane dehydrogenation,the presence of the Lewis acid–base interaction over metal-oxide surfaces may strengthen the coadsorption of a pair of amphoteric species at the M–O site,resulting in distinctly different chemisorption energy and transition state energy scaling relations.As a consequence,the formation energies of H&H coadsorption at the M–O site and H adsorption on top of O were identified as two different reactivity descriptors in the presence and absence of Lewis acid-base interactions,respectively.After the new scaling relations are established in the presence of the Lewis acid-base interactions,the resulting activity plots exhibiting a straight-line,which is very likely the right leg of a new volcanoshaped plot [56].
For the breaking of scaling relations caused by the bifunctional character in heterogeneous catalytic reactions,scaling relations can be rebuilt by identifying electronic or structural properties well suited to describing the various mechanisms or by using binary descriptors to describe the different mechanisms separately.For the bifunctional catalysts in OER,the mechanism changes and the scaling relations for the OER free energy steps are no longer valid.A new scaling relation between the hydrogen adsorption free energy and the highest occupied electronic level aligned with respect to the vacuum level has been re-established [58].The energy level of the highest occupied electronic state can thus be used as a descriptor of the potential of a material to be used as a hydrogen acceptor.For NO oxidation,there are Mars-van Krevelen and Langmuir-Hinshelwood mechanisms on the rutile-type metal oxides (MO2,M=Mn,Ru,Ir,and Rh) at room temperature.Eads(O@M5c) and Ef(Ovac) can serve as two descriptors of the reactivity of the five-coordinated metallic site and the twocoordinated surface lattice O,which give rise to two different oxidation mechanisms.By using binary descriptors,a 3D activity map has been obtained and the optimum activity region for lowconcentration NO oxidation at room temperature has been located.
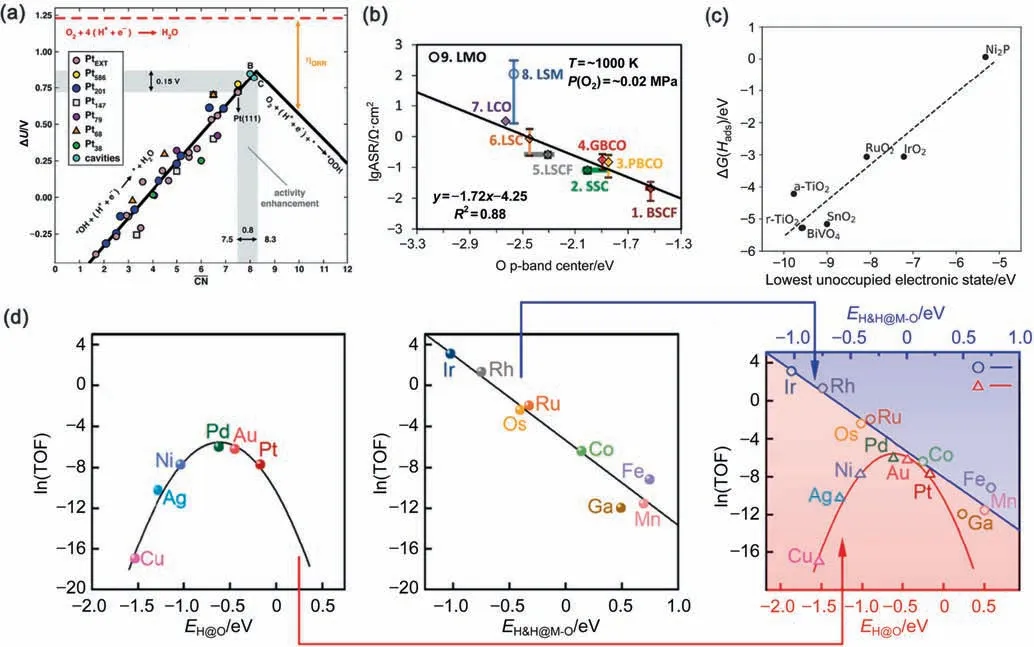
Fig.2.Strategies to rebuild scaling relations (a) Potentials (ΔU) for the two limiting steps on Pt-extended surfaces and nanoparticles and two Pt surface cavities.(b)Experimental area-specific resistances at 1000 K vs.the calculated bulk O p-band center of perovskites with simulated compositions.(c)Free energy of hydrogen adsorption vs.highest occupied electronic level aligned with respect to the vacuum level.(d)Volcano plot of the logarithm of TOF for PDH at 848.15 K,0.02 MPa of C3H8,and 0.016 MPa of H2 as a function of EH@O,EH&H@M–O,and EH@O/EH&H@M–O [56,58,61,62].
5.Establishing Scaling Relations by Machine Learning
Machine learning (ML) techniques have recently successfully accelerated the establishment of scaling relations during the course of catalyst design,making it possible to screen catalysts in a wider variety of catalyst materials.The ways in which machine learning speed up catalyst design can be classified into two main categories,namely,predicting properties such as adsorption energies through electronic and geometrical properties and identifying descriptors and patterns in available kinetic data.
Several studies show that ML can overcome the computational bottleneck of quantum mechanical methods.For example,with the application of Bayesian linear regression (trained using DFTcomputed adsorption energies) and BEP relations,the effects of alloy composition,nanoparticle size,and surface segregation on NO decomposition turnover frequency (TOF) by Rh(1-x)Auxnanoparticles were explored [65].In addition,using Neural Network,training with DFT data,and combining the linear relationship of adsorbates,Li et al.[66] screened more than 1,000 methanol electrooxidation bimetallic alloy catalysts for direct methanol dye cells.On the other hand,non-first-principles features have also been employed to accelerate the selection of catalysts,such as the density and the enthalpy of fusion of metals,which were used in the training of gradient boosting regression to quickly estimate the d-band center for 11 monometallic and 110 bimetallic surfaces [67].These studies show that machine learning methods can help systematically screen the huge catalyst space and provide efficient solutions to complex catalysis problems.
For the identification of descriptors and patterns in catalysis data,various machine learning algorithms such as Subgroup Search and Compressed Sensing have been applied.For example,Goldsmith et al.[68] used subgroup search to classify the crystal phase structure of 82 semiconductors,identified descriptors that can predict the crystal phase structure,and discovered the correlation between the structure of gold clusters and the electronic properties.In addition,the dimensionality reduction algorithm called compressed sensing also provides a powerful and fast method to identify simple descriptors that predict the target properties of materials.In particular,a recently developed algorithm called Sure Independence Screening and Sparsifying Operator (SISSO) identifies low-dimensional descriptors out of a huge feature space (billions of features) within the framework of compressed-sensing based dimensionality reduction [69].For instance,SISSO has been used to search for an improved descriptor to predict the stability of perovskite oxide and halide materials based on an experimental dataset [70].
6.Outlook
Scaling relations and descriptor-based microkinetic modeling have now been widely used to search for new and more effective heterogeneous catalysts.On the one hand,the complexity of catalytic reaction systems leads to the difficulty of building universal scaling relations to identify general reactivity descriptors.On the other hand,it is sometimes desired to break the known scaling relations so as to discover even better catalyst candidates than those lying near the summit of the resulting volcano curve.However,despite a few studies,it is still not clear how to establish new scaling relations to identify new descriptors at a higher level so that they can be applied to a wider variety of catalyst materials.Recently,the application of machine-learning techniques in the field of heterogeneous catalysis has enabled more efficient catalyst design.The machine-learning method can be used to predict structures and energetics according to readily available electronic and geometrical properties and to identify descriptors based on the derived data [68,70–75].Hence,establishing machine-learning models that are highly automated and interpretable non-blackbox would be an effective way to develop generalized scaling relations and to accelerate the identification of universal reactivity descriptors.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (21473053,91645122,and 22073027),the Natural Science Foundation of Shanghai (20ZR1415800),and the Fundamental Research Funds for the Central Universities(222201718003).
杂志排行

Chinese Journal of Chemical Engineering的其它文章
- Struggling as in past, write a glorious future together-CJChE’s 40th anniversary
- Reduced graphene oxide modified melamine sponges filling withparaffin for efficient solar-thermal conversion and heat management
- Photocatalytic degradation of tetracycline hydrochloride with visible light-responsive bismuth tungstate/conjugated microporous polymer
- Ag nanoparticles anchored on MIL-100/nickel foam nanosheets as an electrocatalyst for efficient oxygen evolution reaction performance
- Performance improvement of ultra-low Pt proton exchange membrane fuel cell by catalyst layer structure optimization
- Anodic process of stibnite in slurry electrolysis:The direct collision oxidation