Green biomanufacturing promoted by automatic retrobiosynthesis planning and computational enzyme design
2022-03-01ZihengCuiShidingZhangShengyuZhangBiqiangChenYushanZhuTianweiTan
Ziheng Cui,Shiding Zhang,Shengyu Zhang,Biqiang Chen,Yushan Zhu,Tianwei Tan
National Energy R&D Center for Biorefinery,College of Life Science and Technology,Beijing University of Chemical Technology,Beijing 100029,China
Keywords:Biomanufacturing Retrobiosynthesis Computational enzyme design Biobased chemicals
ABSTRACT Biomanufacturing,which uses renewable resources as raw materials and uses biological processes to produce energy and chemicals,has long been regarded as a production model that replaces the unsustainable fossil economy.The construction of non-natural and efficient biosynthesis routes of chemicals is an important goal of green biomanufacturing.Traditional methods that rely on experience are difficult to support the realization of this goal.However,with the rapid development of information technology,the intelligence of biomanufacturing has brought hope to achieve this goal.Retrobiosynthesis and computational enzyme design,as two of the main technologies in intelligent biomanufacturing,have developed rapidly in recent years and have made great achievements and some representative works have demonstrated the great value that the integration of the two fields may bring.To achieve the final integration of the two fields,it is necessary to examine the information,methods and tools from a bird’s-eye view,and to find a feasible idea and solution for establishing a connection point.For this purpose,this article briefly reviewed the main ideas,methods and tools of the two fields,and put forward views on how to achieve the integration of the two fields.
1.Introduction
The huge value chain extended by fossil resources covers all aspects of modern social life [1].However,the gradual depletion of fossil resources and the global climate problems force researchers and producers to find an alternative development option.Green biomanufacturing,which uses renewable resources as raw materials and bioprocesses to obtain energy and materials,is one of the solutions to achieve sustainable production [2–4].Although the fine chemical industry took the lead in widely applying biocatalysis as an industrial synthesis technology[5],the production of bulk chemicals with an annual output of above 100,000 tons is still dominated by chemical catalytic methods[6].One of the main reasons why biomanufacturing still cannot be fully deployed in the engineering field is the low efficiency of natural synthetic pathways or even lack of biosynthetic pathways for many compounds.There are still some key gaps in their synthetic routes [7] and two main issues are included here:reasonable pathway design and key enzyme design.This makes the whole problem like a road planning problem.If the target compound is imagined as a destination,then the aim of the researchers is planning and finally constructing a’smart road’from an ideal start point,in which the planning corresponds to the design of enzyme-catalyzed cascade pathways,and constructing corresponds to the part of enzyme design (Fig.1).
Just as an overall plan must be carried out before building a road to evaluate the difficulty and rationality of the project,designing an effective biosynthetic pathway is the first step to achieve the biosynthesis of the target compound.This makes it vital to find biosynthetic pathways with complete cascade-connected enzyme catalysis and a referential way to achieve this aim is computeraided retrosynthetic prediction [8].This gave birth to the concept of retrobiosynthesis[9],which similar concepts have been successfully applied in the field of organic chemistry synthesis [10,11].Especially with the rapid progress of deep learning methods in recent years,a large number of significant research results[12,13]and retrosynthetic pathway design tools[14–16]of organic chemistry have been made public.These advanced technologies in organic chemistry also drive the development of methods and tools for retrobiosynthesis.Computer-aided tools represented by Retro-BioCat began to be used to design the enzymatic cascade process[17–19].But limited by the substrate specificity of enzyme catalysis which is different from chemical catalysis,the diversity and effectiveness (or successfulness) of biocatalytic cascade pathway design are often difficult to balance and the number of successful cases of biocatalytic cascade pathways based entirely on computational design is limited.
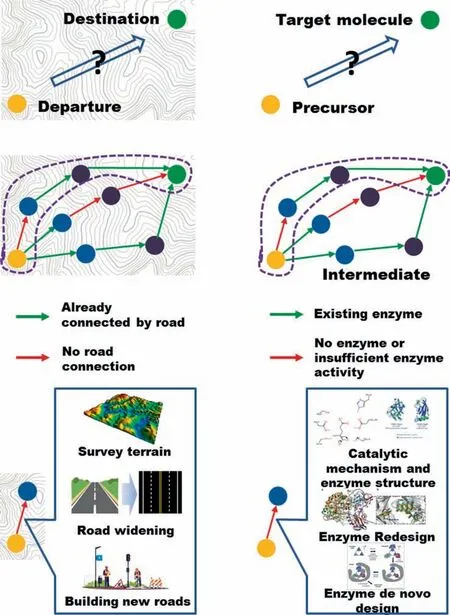
Fig.1.Correspondence between retrobiosynthesis based on enzyme design and road construction problems.
The main cause of this problem is the lack of one or more steps of enzymes,and the computational design of enzymes seems to provide a promising solution.Successful enzyme design will help broaden the boundaries and limits of metabolism and provide more opportunities for synthesis.Many new enzymes with important applications in medicine,industry and research have been created through computational design and related computing tools have also made great strides in the past five years [20].There are mainly two types of computational enzyme design work:redesign and de novo design [21,22].To give a vivid example to reflect the difference between the two types of work,redesign is like widening a road which already exists that can connect two points but not traffic well,while de novo design represent building a road that has never existed between two points.But both of these two types of work are necessary to have a sufficient understanding of the ’construction environment’-catalytic mechanism and protein structure.The acquisition of protein structure and the analysis of the catalytic mechanism have improved our understanding of proteins and provided a basis and a new perspective for the design of functional enzymes,helping us to design or evolve with high activity,environmental tolerance,specific catalytic ability and stereo/enantiomers selective enzymes [21,23–25].
Taking a joint look at the independent development of computer-aided retrobiosynthesis and computational enzyme design,some successful cases of each field seem to indicate that the initial realization of total biosynthesis is not far ahead [26–29].Both methods and tools in the two fields provide users with a wealth of choices.But the core issue—a method,tool,or idea that bridges the two fields—is still unclear.At the level of enzymes,the results of retrobiosynthesis can often only relate to the sequence of known enzymes and the reactions that these enzymes can catalyze.The number of enzymes that have the catalytic mechanism and the exact structure of the active site or even the 3D structure is extremely limited [30].In addition,nothing is known about the enzymatic catalytic mechanism of the new reaction.Unfortunately,this information is necessary for computational enzyme design.If such important information is manually obtained for each individual retrobiosynthesis result,the efficiency of the whole computeraided biomanufacturing will undoubtedly be greatly reduced,and it may also go in the wrong direction when evaluating the calculation method.
But in any case,the existing problems did not make researchers in the two fields stop their respective research steps.Methods and tools are still being reported constantly and related reviews are endless.The foundation required to build a connection point between these two fields is also not completely blank.To achieve the final integration of the two fields,it is necessary to examine the information,methods and tools from a bird’s-eye view,and to find a feasible idea and solution for establishing a connection point.For this purpose,we try to review and briefly explain the related studies that have been published in this review.This review will be described in two parts include retrobiosynthesis (enzyme cascade catalytic pathway design) and computational enzyme design,and will discuss the direction and plan of how to establish a strong relationship between the two fields at the junction.Since there is no lack of excellent and in-depth reviews in a single field,we do not intend to elaborate on specific technical details in this review,but try to present the blueprint for future computeraided biosynthetic and biomanufacturing that these reviews can produce to readers by fusing them together [21,31–37].
2.Retrobiosynthesis
The tremendous progress in sequencing technology,metabolomics,proteomics and related detection technology in recent years has made it possible to build large metabolic databases,and the ability to mine these databases has led to the idea of automating the process of finding alternative and novo pathways with different enzymes by using retrobiosynthesis.Retrobiosynthesis works reversely through biochemical transformation to find possible enzyme combinations to complete the production of target molecules from natural metabolites [38].
The minimum information required to implement the retrobiosynthesis algorithm is the initial set of precursors and reaction rules.Considering that the list of metabolites that can be used as precursors and can be produced by microbial metabolism is always easy to obtain and consistent in different studies,constructing and making a set of reaction rules becomes the basis for retrobiosynthesis.Once the reaction rules set is obtained,retrobiosynthesis is generally divided into two parts:retrobiosynthesis pathway enumeration and pathway evaluation.
2.1.Constructing reaction rules set and retrobiosynthesis pathway enumeration
Retrobiosynthesis was originally developed for chemical synthesis,which involves mining the chemical reaction network from the target backward until the desired precursor is reached.Similar to the retrosynthetic,the generation of the retrobiosynthesis network is based on the basic assumption that similar substrates are more likely to undergo similar enzymatic reactions.The construction of a retrobiosynthesis network requires a digital representation of chemical molecular formulas and reactions.The most common representation method is based on reaction rules generated by mining and classification of known biological and chemical reaction databases.
With the accumulation of knowledge,a large number of biological and chemical reactions are stored and recorded in computerrecognizable coding forms.A large number of biological and chemical reaction databases such as Reaxys [39],SciFinder [40],ChemSpider[41],SPRESI[42],KEGG[43],MetaNetX[44]and Rhea[45] provides the ability to explore the synthesis space of new compounds.
Whether it is chemical synthesis or biosynthesis,the vast majority of computer-aided retrosynthetic programs rely on encoding reaction rules (templates) or generalized subgraph matching rules [46].The reaction rule mentioned here refers to the chemical reaction represented by the molecular fragments of reactants and products within a certain radius around the reaction core[47].The general reaction rule extraction includes the following steps:
(1) Use automated tools or manually to map the reactants and products of each reaction in the chemical reaction data set to establish the atomic changes during the reaction;
(2) Extract the atoms and bonds in the reaction center and its surrounding environment according to certain rules for each chemical reaction in the reaction data set,and obtain the reaction rules for each reaction;
(3) Classify the rules obtained,and the reactions with the same reaction rules are classified as a similar reaction
The reaction center here refers to a group of atoms in which the atoms’ connection situation or related bonds change from reactant to product.A rule-based approach requires a decision on the degree of generalization.The more general the rule is,the fewer atoms it contains,and the more similar reactions it can represent.Conversely,the more specific the rules,the more structural details it contains,and the fewer responses it can represent.Fig.2 shows the general process of rules (templates)extraction.Based on this process,many retrosynthesis or retrobiosynthesis software or methods have established their own different rule sets [47–51].
The degree of generalization of the reaction rule depends on the diameter defined around the reaction center.As the diameter of the reaction rule becomes smaller,more substrates gather together as equivalent classes in the metabolic space,representing the same degree of confounding of the conversion.Although a larger radius can be closer to the structure of a known active substrate,it will lead to a decrease in the generality of the rules.In addition,the complexity of the enzyme catalytic mechanism makes the similarity of substrates not completely correlated with enzyme catalytic activity[52].Nevertheless,the similarity of the substrate structure is still the main indicator for evaluating the feasibility of the listed pathways in the results of retrobiosynthesis.
According to the rule set obtained,a tree-like result representing the potential synthesis path of the target molecule can be enumerated.The depth of this tree is generally specified manually and the number of branches at each node is related to the size of the rules set.Small and generalized rules set can enumerate the possible reaction precursors more comprehensively,but it will cause each node to have a larger number of branches,resulting in a combinatorial explosion.As shown in the work of Mathilde Koch et al.[53],the number of branches per node expands from 100 to greater than 1000 with the expansion of the rules set,and a significant increase with the addition of the rules which have a low radius and high confoundness.
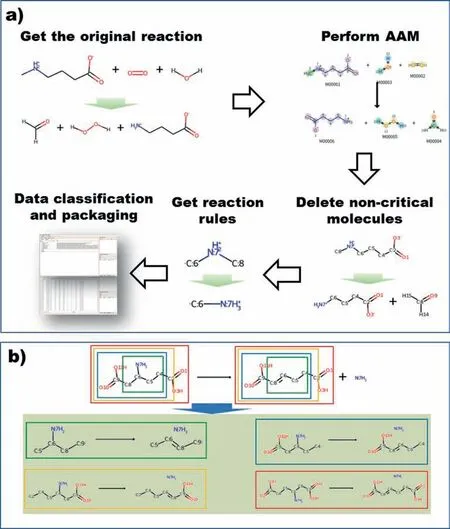
Fig.2.(a) The process of obtaining reaction rules and (b) the rules of the same reaction under different diameters.(Different color boxes represent different diameters).
A very important process in the extraction of reaction rules is to establish a precise Atom-Atom Mapping (AAM),which means the correspondence between the substrate and the product atoms.The AAM process determines which bonds are broken/formed,or which bonds change their order [54].The main methods used for AAM are Maximum Common Structure (MCS) and Mixed-Integer Linear Programming (MILP).Table 1 lists four widely used automatic AAM tools and their features[55,56].Nuno Oso’rio et al.evaluated the effects of these four tools.The results on the data set compositions by 11,575 different reactions show that up to 95%of the effective AAM can be achieved in the RDT mapping results[57].

Table 1 Main Atom-Atom Mapping tools and their Features
2.2.Retrobiosynthesis pathways evaluation
The main goal of retrobiosynthesis is to discover a valued de novo pathway since its concept was introduced [26,58].However,the retrobiosynthesis enumeration algorithm can generate a large number of hypothetical pathways based on reaction rules without considering the enzyme,genetic regulation and host background information required for the implementation of the actual metabolic pathway.This makes a large number of hypothetical pathways present false positives,and it is difficult to bring actual results.Therefore,an algorithm or method is needed to evaluate and screen hypothetical pathways obtained by enumeration.
The main evaluation indicators for a retrobiosynthesis pathway include enzyme performance,reaction similarity,pathway length,thermodynamics,toxicity,yield,etc [59].Among them,the performance of the enzyme is the most important indicator,and it is directly related to the realization difficulty of the pathway in the experimental.In addition,the length of the metabolic pathway is also an important indicator.In the case of enzymatic activity in each step,the excessively long pathway length directly affects the metabolic burden of the host cell and the product yield.Indicators such as thermodynamics and toxicity are also part of the evaluation.This is mainly to evaluate the feasibility of the direction of the reaction and flow distribution,as well as the impact of metabolites on the growth of host cells.Usually,these factors will be considered in the form of a scoring function and give a score for each pathway [60].
Although the scoring function can cover common evaluation indicators,it is often not consistent with the score in the actual selection of the best pathway that is valued to carry out in the next step.The intuition of scientists is still a non-negligible influence.For such a vague and complex problem,artificial intelligence strategies seem to provide a solution and have been successfully used in chemical synthesis and metabolic engineering[14,16,51,53].Due to the molecular fingerprints are usually represented by a set of vectors and it is convenient to define the distance,a simple idea is to use machine learning to establish the relationship between the molecular fingerprints of the substrate and the product,so as to realize the positioning of the pathway retrieval direction[14,51].The other is the contemporary‘artificial intelligence’ based method that emerged with the success of AlphaGo [61,62].Since the tree-like decision strategy making of retrosynthetic itself has the same effect as the decision of Go,the machine learning method in chess confrontation also shows value in the field of retrobiosynthesis.Monte Carlo Tree Search (MCTS)reinforcement learning method is a representative algorithm[16,53].Its main advantage is that it can balance the exploration of the unknown space and the use of existing knowledge.
Based on the above principles and strategies,some tools for enumerating and evaluating retrobiosynthesis have been developed.The current main retrobiosynthesis tools are mainly shown in Table 2.Among them,as an early framework,BNICE [63] is based on a manually planned reaction rule set to reflect the best possible knowledge of enzyme confounding.However,manually adding rules is difficult to adapt to the constant discovery of new reactions.In order to adapt to this change,the rules must be automatically extracted from the database.The typical representative of this is RetroRules and the pathway search tool RetroPath based on it[64].RetroRules provides a complete set of reaction rules,covering more than 16,000 kinds of biochemical transformations expressed in varying degrees of confounding and covering stereochemistry.In addition,there are some similar works.The reaction rules of the MetRxn database are calculated by the code available on GitHub,but its rules database is currently not available [65].MINE,a database that uses BNICE reaction rules to generate new chemical structures for metabolomics data analysis,but does not provide the details of reaction rules [66].Its latest work has updated the rules set,and the general rules have reached 1224[67].The newly published RetroBioCat,as an enzyme cascadecatalysis design toolbox with an intuitive visual interface,uses a rules set consisting of 99 reactions [17].
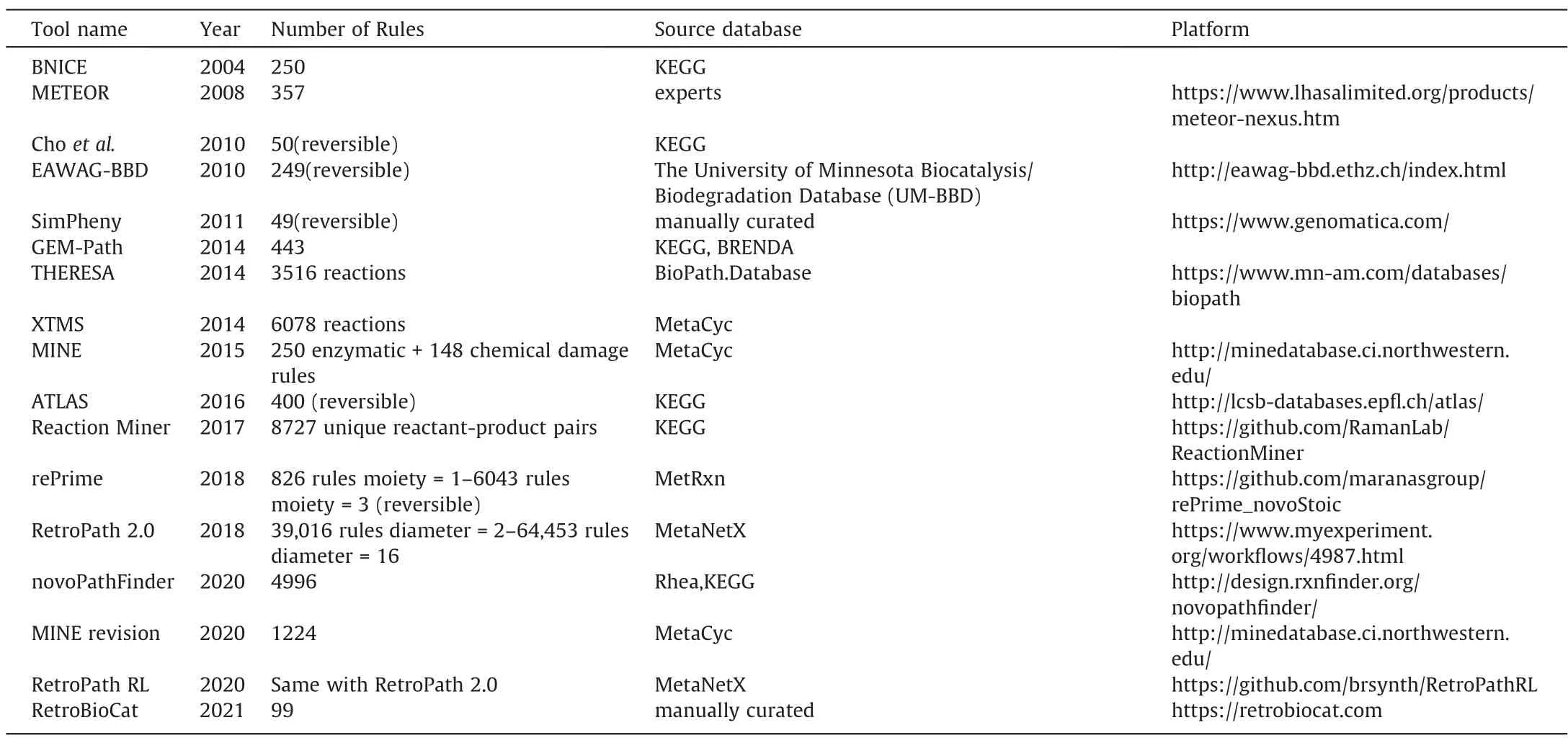
Table 2 Main automatic retrobiosynthesis tools
Although these tools have not yet been widely used and accepted by experimenters,some tools still perform well in practical applications.Here some typical and latest application cases are briefly explained.Based on SimPheny,Harry Yim et al.[26] gave a variety of pathways for the biosynthesis of 1,4-butanediol (BDO)from common metabolic intermediates and designed an E.coli host to achieve a direct biocatalytic route from renewable carbohydrate raw materials to BDO which the output reached 18 g·L-1.Zhenning Liu et al.[68] constructed an artificial 3-phenylpropanol pathway based on RetroPath 2.0 and achieved the highest titer production in E.coli.Based on BNICE.ch,Jasmin Hafner [69] and others designed and realized a new biosynthetic pathway of (S)-tetrahydropalmatine and its derivatives.Liu et al.[70] used EWAG-BBD proposed a D-xylonate-dependent pathway and demonstrated the first example of ethylene glycol(EG) biosynthesis from pentose xylose by engineered E.coli.In addition,there are some successful applications based on BNICE,RetroPath and other tools in biosynthesis pathway design of terpenes,ethylene glycol,levulinic acid,propionic acid,butanol [71–75].These successful application cases all demonstrate the potential of retrobiosynthesis in biomanufacturing.
2.3.Retrobiosynthesis planning motivated by enzyme design
Although retrobiosynthesis has shown great development prospects and a large number of new synthetic pathways “designed” by computers make people feel that the milestone of achieving “total biosynthesis” is not far ahead,but the road underfoot does not extend forward anymore right now.The five levels of metabolic engineering proposed by Tobias J Erb et al.[76]can well reflect this problem.Among them,the 1,2 and 3 levels of metabolic engineering all rely on known enzymes which represent what synthetic biology is mainly doing now.But in the 4 and 5 levels where retrobiosynthesis can give full play to its advantages,the lack of knowledge of new enzymes and the reactions they can catalyze makes the progress of synthetic biology difficult.But some work based on computational enzyme design has provided the light for overcoming this obstacle [27–29].Some enzymes with new synthesis capabilities have been designed and prepared,which making some compounds have completely unnatural biosynthetic pathways.This means new enzymes created by enzyme engineering or de novo design can pave the way for real total biosynthesis.
3.Computational Enzyme Design
Enzymes as catalysts can sometimes enhance the reaction rates by more than 10 orders of magnitude [77].Natural enzymes are the result of long-term evolution in nature,and their extremely high substrate selectivity also limits their scope of application[78].The high specificity of enzymes results in that it is not always possible to find specific enzymes that catalyze reactions with industrial value in nature,and harsh reaction conditions also increase the difficulty of industrial production [79].Therefore,it is necessary to design and modify the enzyme to overcome its limitations so that it can be better used in industrial production.An indepth understanding of the enzyme reaction is very important for the successful construction of any characteristic of the enzyme(whether it is stability or specificity).Enzyme design usually focuses on the change of reaction energy barrier between the enzyme-substrate complex and transition state.The general design concept is to enhance the catalytic ability by constructing and reducing the total energy of the transition state or the enzymesubstrate complex that is geometrically close to the transition state[80,81].Computational enzyme design is a promising technology,that is structure-based computational protein design (CPD)method used to construct new enzymes[82,83],and new catalytic reaction performance can be rationally designed according to needs.CPD has been developed for 20 years to assist specific scene sequences to adopt specific three-dimensional structures and have the required characteristics [84,85].
Most CPD algorithms consist of three steps:(i) side chain generation step,generating discrete side chain rotamers at predetermined positions on the protein backbone template [86],(ii)energy calculation,where the potential energy function Used to calculate the interaction energy between each rotamer and the framework template [87–89],and (iii) sequence optimization steps,in which the search algorithm is used to optimize the combination of rotamers [90–95] and explore sequence space with rotamers to determine the best sequence.After that,a series of sequences and their corresponding rotamer optimized structures will be generated,and each sequence will be sorted according to the energy reflecting its predicted stability of the target protein structure.
According to the degree of enzyme modification,computational enzyme design can be divided into the redesign and de novo design(Schematic of design method was shown in Fig.3).Enzyme redesign refers to changing certain physical and chemical properties of natural enzymes that already have the ability to catalyze target reactions while retaining the original catalytic sites to make them suitable for industrial reaction conditions.The de novo design requires the artificial construction of active centers,so that the inert protein backbone has the ability to catalyze the target reaction.Regardless of the design method,it is necessary to create a binding pocket specific to the required substrate,introduce the required catalytic mechanism for the chemical reaction of interest,and combine one or more catalytic side chains with high energy reaction intermediates are limited to the corresponding state according to a set of predefined geometric constraints.To understand the transition state structure,quantum mechanics is needed to analyze the catalytic mechanism,or directly use the knowledge recorded in the existing catalytic database.In addition,the computational enzyme design is based on the three-dimensional structure of the enzyme.The design becomes particularly difficult when the crystal structure is lacking.Therefore,numerous researches on protein modeling have also been carried out.In the following subsections,we will discuss in detail the key technologies involved in the computational enzyme design.
3.1.Protein structure and prediction methods
Computational enzyme design relies heavily on precisely resolved protein structures.If the structure is missing,there is no way to calculate the enzyme design.Advances in software and automation have accelerated the growth of the experimentally determined protein structure database (Protein Data Bank (PDB))[96],which now contains nearly 180,000 macromolecular structures.However,the number of known protein sequences is much larger than the number of resolved protein structures,and relying solely on the inefficient experimental determination of protein structures is a drop in the bucket for the vast protein sequence database.
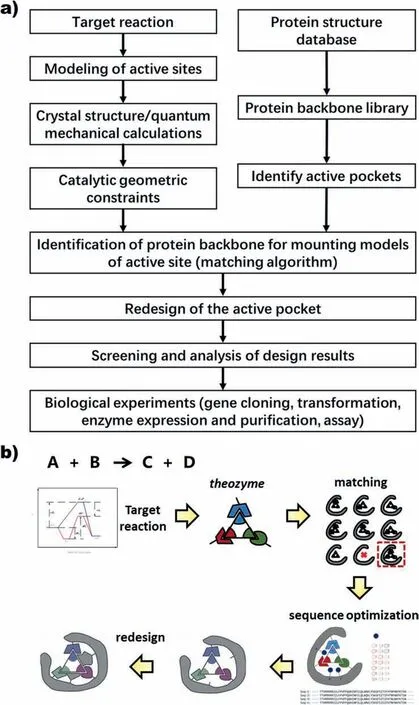
Fig.3.(a) Flow chart of computational enzyme design;(b) Schematic diagram for de novo enzyme design.
Considering the difficulty of current experimental measurements and the speed of discovery of new protein sequences,it is important to find alternative methods to determine protein structures.Protein structure prediction methods infer the threedimensional structure of a protein from its amino acid sequence.Based on the availability of template structures in protein structure databases such as PDB,methods for computational structure prediction can be divided into three categories:homology modeling based on homologous structures [97],threading algorithms based on existing folded modules [98,99] and template-free methods(previously called ab initio structure prediction) [100–103].The tools commonly used in each method are listed in Table 3.Among them,homology modeling finds proteins with high sequence similarity and resolved structures as modeling templates,which requires high sequence similarity of the templates,and when the sequence similarity is low(<30%),it is difficult to perform accurate homology modeling.SWISS-MODEL [114,115] is a well-known online tool developed by Torsten Schwede’s structural bioinfor-matics group.Fast operation,simple operability and rich functionality have made it one of the most widely used homology modeling tools of the last 20 years.The threading algorithm does not require highly homologous structures,but it does require smaller folding modules from existing protein crystal structure data.I-TASSER[116],developed by Zhang’s group,based on the threading algorithm and fold recognition to obtain the final structure by template identification,assembly and overall structure optimization.It has remained a leader in the last decade of CASP.This was followed by the development of C-I-TASSER,which integrates deep learning of interresidue contact maps and I-TASSER to create correct folding for 85% of the proteins in the SARS-CoV-2 genome.When the template-free method is required,there is essentially no available template to use as a reference,and it is generally possible to accurately predict the structure of proteins of small length (less than 200 amino acid residues).Rosetta [117] is the first template-free method developed by David Baker’s lab that assembles structures based on fragments of 3–9 residues in the PDB structure.QUARK[118] is another excellent fragment assembly method developed in the laboratory of Zhang Yang.There are also many templatefree methods,such as PROFESY [119],FRAGFOLD [120],and so on.These methods do not rely on any global structural template and can be more capable of modeling newly folded targets.However,it is still a big challenge for template-free.
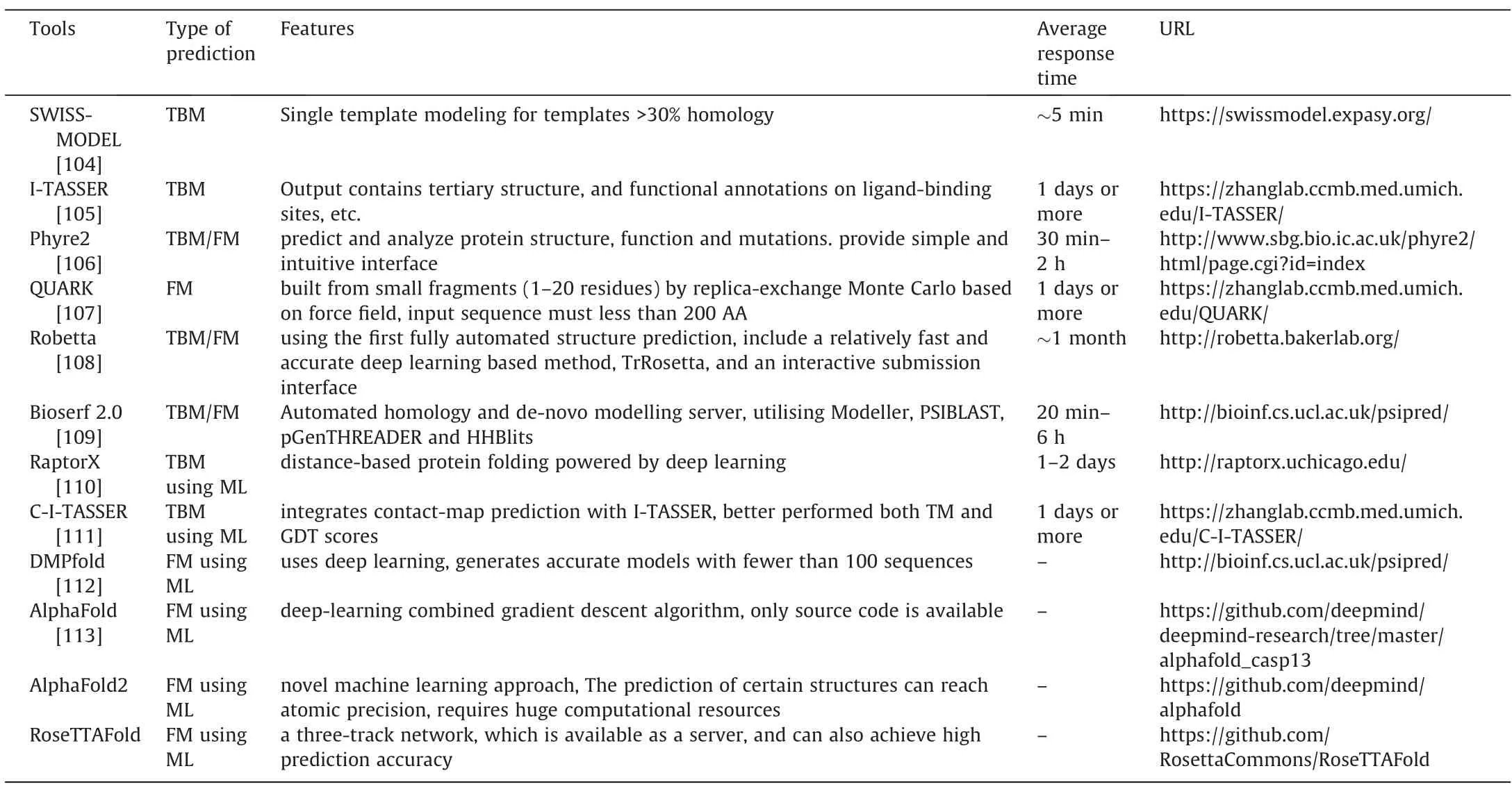
Table 3 Commonly used tools for protein structure prediction
CASP [121] (critical assessment of structure prediction) is a two-year community experiment designed to evaluate,promote and confirm the best means of protein structure prediction.CASP13 has made significant progress in structural modeling by applying deep learning techniques to successfully predict inter-residue distances without the use of structural templates.In the latest CASP14,AlphaFold 2,from DeepMind,shines again,which entry achieved a median score of about 92%GDT_TS(while models with values greater than 75%have many correct atomic-level details),a measure of amino acid residues being within an acceptable accuracy threshold of experimentally determined positions across more than 90 structure predictions.AlphaFold2[122]is a novel machine learning method that includes a variety of highly sophisticated components.One of these components,Evoformer,can effectively extract information from multiple sequence alignments and establish an accurate representation of the part of the protein in close contact.The Structure Module creates a three-dimensional structure for the protein.Coupled with a lot of deep learning skills,the model can produce surprisingly accurate predictions.In addition,they applied AlphaFold2 to 98.5% of the human proteome[123].The resulting data set covers nearly 60% of the amino acid structure predictions of the human proteome,and the prediction results are reliable.The predicted information will be freely available to the community through a public database hosted by the European Bioinformatics Institute (https://alphafold.ebi.ac.uk/).At the same time,the Baker team put forward a 3-track attention mechanism through research on the network architecture combined with related ideas [124].One-dimensional sequence-level information,two-dimensional distance map-level information,and three-dimensional coordinate-level information are successively transformed and integrated to make the entire neural network learn information of three levels at the same time,to obtain the best network performance.Its newly developed RoseTTAFold not only has an accuracy close to the prediction accuracy of DeepMind in CASP14,but also surpasses the calculation speed and computational power requirements.This method is also available as a server at https://robetta.bakerlab.org (RoseTTAFold option).Such significant advances by end-to-end difference systems consisting of multiple neural network modules successfully predicting not only three-dimensional contacts but also more general interresidual distance distributions have generated great interest and creative thinking,and breakthroughs in protein structure prediction problems will yield huge leaps forward for enzyme design.
3.2.Resolution of the catalytic mechanism
Just as the geological characteristics of the construction site need to be investigated in detail before the actual construction,the resolution of the catalytic mechanism is important for the successful modification and design of the key characteristics of the enzyme -catalytic efficiency,substrate specificity,stability.The main point of mechanism resolution,however,is the energy distribution of the reaction process,specifically targeting the energy difference between the enzyme-substrate complex and its transition state.Enzymes can be designed by reducing the total energy of the transition state or of an enzyme-substrate complex that is closer in energy and geometry to the transition state [81,125,126].Four methods commonly used to resolve this energetic process are summarized below:the quantum mechanical cluster approach[127],hybrid methods using quantum mechanics/molecular mechanics,empirical valence bonding (EVB),and hybrid models using quantum mechanics/molecular mechanics/molecular dynamics.A comparison of the advantages and disadvantages of the four methods is summarized in Table 4.
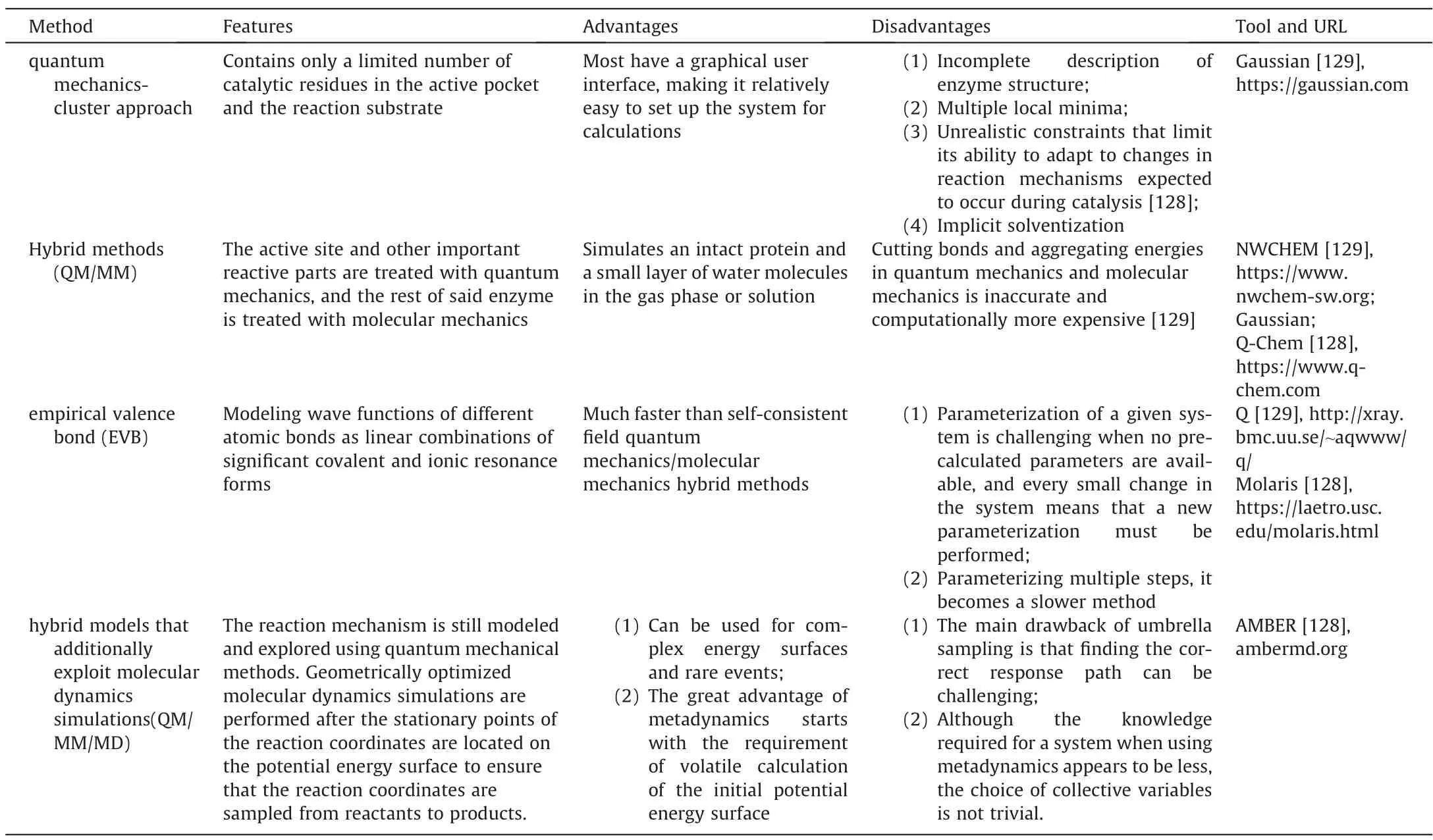
Table 4 Commonly used tools for the calculation of potential
3.3.Catalytic mechanism-related databases
Although several quantum chemical calculations have been developed,finding the correct transition state and its internal reaction coordinates is still a difficult and lengthy task.The difficulty of mechanism resolution and the importance of modification for enzyme design have led to the advancement of the development of relevant databases for known enzyme catalytic mechanisms.
The M-CSA [30],as a representative mechanism database,was created to capture and organize these and other types of mechanism data available in the literature and make them available in a community format in a standardized and computer-readable form.The M-CSA includes the complete catalytic reaction as well as annotations for each step of that reaction,which include a curly-arrow description of the step-by-step chemical reaction mechanism,the role of each catalytic residue and any cofactors,as well as supporting data from the primary literature.As of June 23,2021,the M-CSA contains 964 hand-curated entries,684 of which have detailed mechanical descriptions.These represent 852 EC numbers.There are 73,158 SwissProt sequences and 15,487 PDB structures homologous to these entries,which catalyze the same reactions and have the same active sites,and thus may follow the same mechanism.
The EzCatDB database [130] (http://ezcatdb.cbrc.jp/EzCatDB/)emphasizes the manual classification of enzyme reactions in terms of enzyme active site structure and their catalytic mechanism based on literature,amino acid sequences of enzymes(UniProtKB)and the corresponding tertiary structure of the Protein Data Bank(PDB) and currently contains 880 entries.The database includes information related to ligand molecules on enzyme structures in the PDB data,classified according to cofactors,substrates,products and intermediates,which are necessary to elucidate the catalytic mechanism.
MACiE [131] (Mechanism,Annotation and Classification in Enzymes) is a database of enzyme reaction mechanisms that can be accessed at http://www.ebi.ac.uk/thornton-srv/databases/MACiE/.It includes detailed step-by-step mechanistic information in a wide range of chemical spaces and protein structures to provide computational descriptions of mechanisms.This version of MACiE represents the addition of 133 new entries since the last major release (bringing the total number of entries to 335).It now covers over 90%(182)of the EC subclasses with available crystal structures,representing 321 different EC numbers.
Although quantum mechanical methods are the most direct means of understanding reaction mechanisms,the problem of computational over-expensiveness is always unavoidable.Database access to the classification of catalytic mechanisms and anno-tation of ligands allows detailed elucidation of the relationship between the active site structure and function of enzymes,thus eliminating the need for literature searches and computational processes.However,the existing database of catalytic mechanisms still suffers from a small amount of data and the problem that possible catalytic residues have been identified but the complete mechanism is not yet clear,and additions to the database require more efforts from researchers and possibly the use of additional protein structure tools to improve the database.
3.4.Redesign of enzyme
For natural enzymes that can catalyze the target reaction type and have some residual hybrid activities,retaining their original catalytic mechanism and catalytic sites,optimizing the amino acid sequence and screening to obtain enzyme mutants with corresponding functions can be regarded as a redesign [132].The general workflow is:based on the properties of the substrate of interest and the transition state calculated from quantum mechanics (QM),the transition state structure is placed in the active pocket according to the predetermined catalytic geometric constraints.This can be done using the ligand placement algorithm[133].Once the location of the catalytic residue is determined,the amino acids adjacent to the active site can be designed to optimize substrate recognition and transition state energy barriers.
It is expected to obtain an optimal sequence structure when designing a protein,The physical meaning of the optimal sequence is often clear,that is,it has high catalytic activity and strong stability,etc.,but the quantitative criteria for the optimized sequence are difficult to determine.The accuracy of full sequence optimization affects the final result,so a variety of algorithms have been developed for sequence optimization.For the use of Orbit [133],the residues needed to accommodate substrate binding are determined by minimizing the total energy of the system.Pairwise force fields [134] and dead-end elimination [135] are used in protein design based on combinatorial optimization algorithms.Rosetta-Design also uses the total energy of the system as the objective function of sequence optimization,but the interaction energy between the transition state and the protein side-chain is increased,and the force field used depends on the environment,which limits the use of deterministic algorithms in sequence selection.Therefore,random optimization algorithms such as Monte Carlo are used in RosettaDesign [136,137].ProdaDesign [95] is a deterministic optimization algorithm based on DEE/LP/MILP[138] proposed by Huang et al.It can obtain the global optimal solution of energy in large-scale amino acid sequence combination problems.So that the active site can be better pre-organized and can be combined with the substrate in the correct catalytic mode.A few computational design tools with successful design examples have been developed and are listed in Table 5.
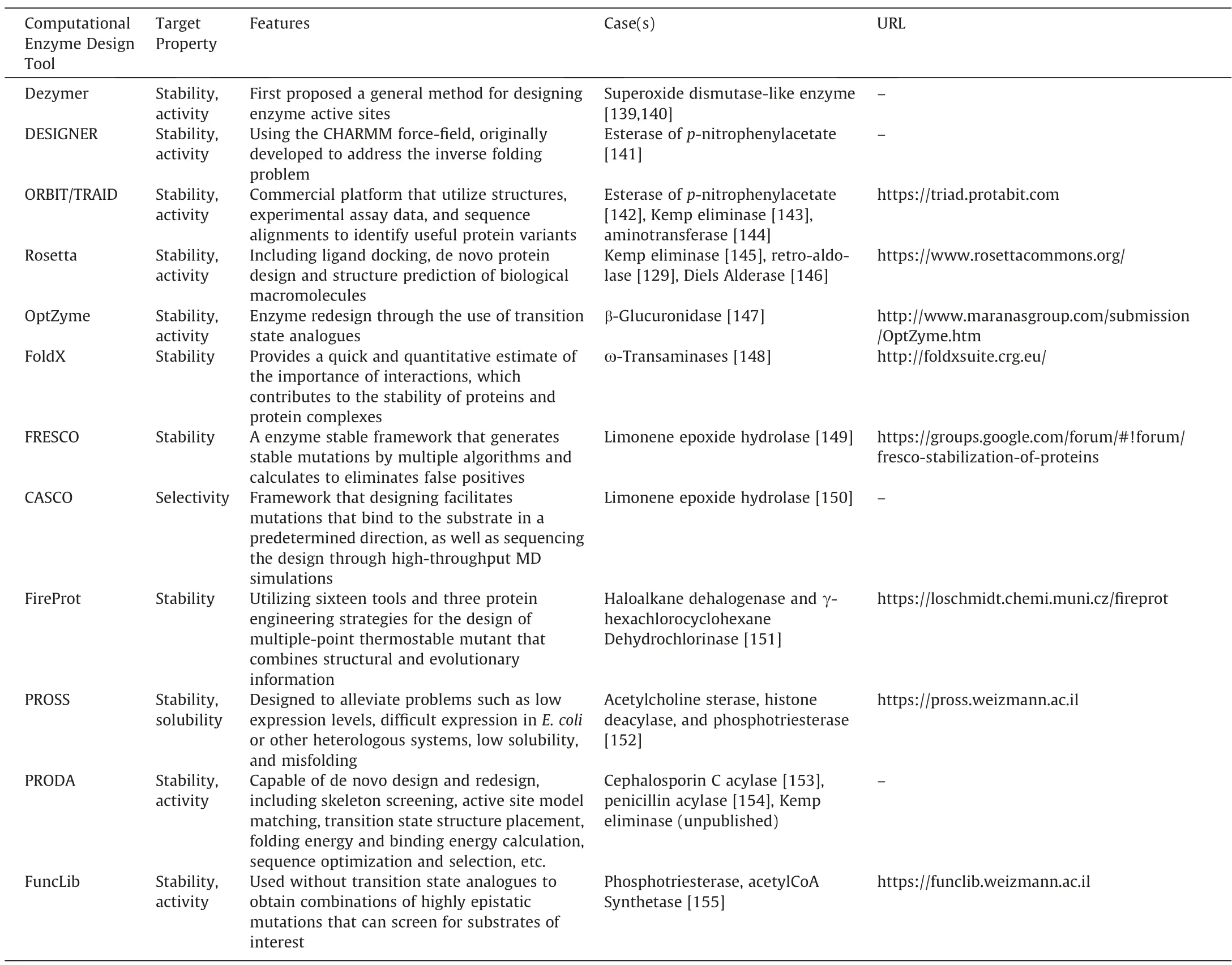
Table 5 Computational enzyme design tools
Although not as aspiring as designing from scratch,the redesign of natural protein scaffolds often represents the quickest path to new catalytic activity.The existing active site can be easily modified by reasonable and random mutations.In some cases,very few mutations are enough to cause significant changes in function[156].The conversion of pyridoxal phosphate-dependent racemase to aldolase through an active site mutation is one example[22].He et al.redesigned penicillin acylase and obtained a penicillin acylase mutant (M142αF/F24βA/S67βA) with high selectivity,whose synthesis/hydrolysis ratio was increased by more than 10 times[155].The yield of the mutant enzyme can reach 99%under industrialized conditions after immobilization.Generally,in other cases,more extensive reshaping may be required.
Utilizing features of the original active site,such as the basebinding pocket and existing functional groups,reduces the number of changes required for the redesign.Thus,the resulting activity is usually related to the original function of the protein,using the existing catalytic mechanism.To obtain more distant activity,the weakly mixed side reactions of the scaffold can sometimes be amplified [156].Wu et al.came to transform a very narrow substrate range enzyme,Bacillus sp.YM55-1 aspartase,with a set of complementary hydrogenated amination biocatalysts [157].The redesigned enzyme catalyzes the asymmetric addition of ammonia to substituted acrylates,providing enantiopure aliphatic,polar,and aromatic β-amino acids.Without further optimization,the redesigned enzyme exhibited substrate tolerance up to 300 g·L-1concentration,up to 99% conversion and selectivity.
Natural cofactors extend the properties of enzymes by facilitating otherwise difficult transformations.Clipping their surroundings or replacing natural cofactors with synthetic alternatives can yield new functions.Heme proteins have been redesigned in this manner [158,159].This approach has recently yielded artificial metalloenzymes for the enantioselective hydrogenation of olefins,transfer hydrogenation reactions of ketones and olefin complexation reactions,among other activities [160–162].Such systems combine the intrinsic reactivity of small molecules with the specificity of proteins to optimize their properties by chemical mutation and directed evolution,respectively.
Mechanistic studies of catalysts and their optimized progeny during redesign have the potential to provide a deeper understanding of enzyme chemistry,thus providing valuable perspectives to study the utilization of binding energies in catalysis and the evolution of biological functions.A better grasp of the sequence-struc ture–function relationships of these proteins could also help optimize design algorithms.However,generating entirely new types of reactions by redesign is much more difficult.
3.5.De novo design of enzyme
The purpose of the de novo design of enzyme is to create a naturally-inspired catalyst that specifically produces the desired compound of interest.New protein design[22]is seen as an opportunity to explore the universe of “protein space”[163].Theoretically,the spatial structure of a protein is entirely determined by its amino acid sequence,and conversely,it is possible to design a specific amino acid sequence so that it can spontaneously fold into a desired three-dimensional structure with certain functions,a process that can be called de novo design of proteins [164].However,the success rate of de novo design of protein structures is low because the combinatorial space of protein sequences is very large and the energy functions used in current modeling methods still suffer from insufficient accuracy.Although the rapid development of structure prediction methods in recent years has helped researchers to quickly pick out the predicted sequences that can really fold into the target structure by high-precision structure prediction,the obtained results can often only approximate the folding into the desired structure [165,166],and their accuracy is not yet sufficient for computational enzyme design.Therefore,the current de novo design of enzymes usually still relies on the natural protein backbone as a starting point to find the desired folding pattern from a large structural database [167,168],on top of which artificial active sites are introduced by mutation,so as to ensure the design accuracy.
First,a theoretical enzyme active site model (namely “theozyme”) of catalysis is constructed for the target reaction[169].After the mechanism of the reaction is clarified,the structure of the transition state,the residues to achieve the catalytic function is determined,and then the geometric constraints required for catalysis are obtained by optimization with the help of quantum chemical methods to simulate the reaction mechanism of the enzyme and stabilize the transition state structure [81].Next,the model is made to anchor on the existing protein scaffold.It is usu-ally necessary to first scan the protein backbone to identify possible active pockets and search for suitable sites in the active pocket to install specific residues.Once the match is completed other amino acid sites around the pocket need to be redesigned,meaning that the entire protein sequence and structure are re-optimized to allow better binding affinity to the transition state through hydrogen bonding,hydrophobic stacking,and other interactions.Although de novo designed biocatalysts sometimes do not perform very well,they can be highly improved by successive rounds of directed evolution [170–172].Enzymes designed from de novo should precisely bind their substrates in a catalytically productive binding mode,increasing the difficulty of generating entirely new activities,yet once a low level of activity is identified,an evolutionary approach is particularly helpful in expanding it.
From small modifications to the active site of enzymes to the de novo design of new catalysts,a growing number of computerbased strategies have been developed to achieve this goal.De novo design has been underway since 1991 when Hellinga and Richards designed their molecular modeling program DEZYMER to provide a general method for designing enzyme active sites[173].Bolon and Mayo extended their successful computational protein design tool,ORBIT [84,174],to the field of enzyme active site design and created a histidine-containing catalyst for the hydrolysis of pnitrophenyl acetate to p-nitrophenol.Later,Baker and colleagues created several artificial enzymes with significant activity for typical bond-breaking or bond-forming reactions by using the design module in the protein modeling software ROSETTA [175,176].Three major important efforts were also truly successful in the de novo design of active enzymes:Kemp elimination [171],the Retro-Aldase reaction [129],in which carbon–carbon bonds are broken in non-natural substrates (i.e.,not found in any biological system),and the Diels-Alder reaction [177].So far there are still few tools that have been successfully used in de novo design,and some examples of de novo design and related software are listed in Table 6.
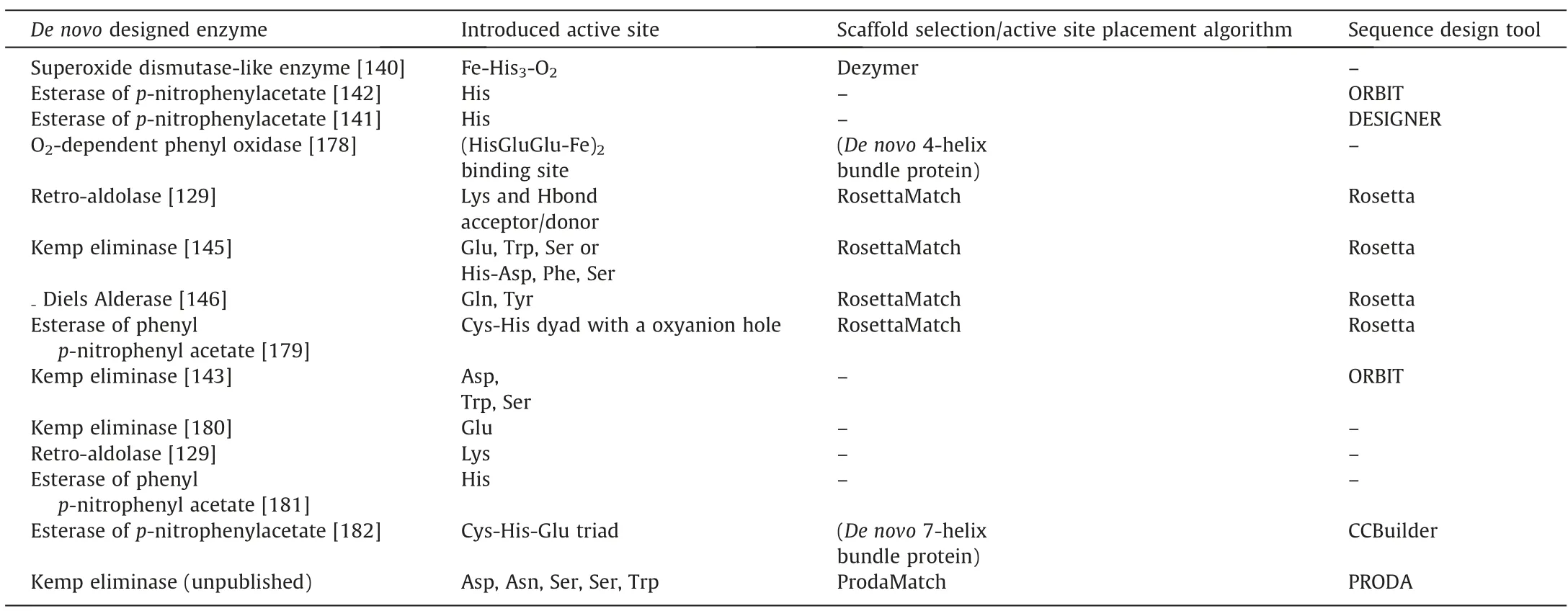
Table 6 Cases of de novo enzyme design and tools
The most critical problem in de novo design focuses on how to introduce the theoretical enzyme model to the protein backbone.The complexity of the process comes from identifying the position of each catalytic residue in the theoretical enzyme and determining its correct conformation to meet the strictly predefined catalytic constraints.A variety of algorithms have been developed by many groups to solve the matching problem of active site models.Matching algorithms including RosettaMatch [175],OptGraft[183],ScaffoldSelection [184],PRODA_MATCH [185–187],Auto-Match [188],Saber [189],and HBNetGen [190] have all demonstrated powerful inert scaffold matching and catalytic residue anchor capabilities.Among them,the matching tool PRODA_-MATCH [185–187] developed by Zhu et al.based on the Quasi-Newton BFGS algorithm and heuristic search algorithm can greatly improve the matching efficiency and accuracy because it does not rely on the side chain rotamer library in the residue installation process but adopts the method of continuous conformational change to determine the conformation of catalytic residues and transition states.It improves the matching efficiency and accuracy,and is able to anchor 8 or even more catalytic residues at one time,thus greatly expanding the application of de novo design methods in more complex enzyme-catalyzed systems.
De novo design has become mainstream,with more than half of all review articles employing this approach to some degree.This unprecedented research activity has made enzyme design more robust,accurate,and reliable [32].Despite the remarkable success that has been achieved,there are still huge differences between natural and designed enzymes (up to 11 orders of magnitude)and indeed the energy landscape and the time scale of conformational changes are also critical for enzyme activity.ludwiczak et al.combined Rosetta with molecular dynamics(MD)to enhance protein structure design and corresponding sequence selection [191].The combined approach produced 20%–30%more sequences than existing methods,improving the diversity of sequence prediction and similarity to natural sequences.The designed interaction network between residues and the transition state structure undergoes continuous dynamic changes through conformational rearrangements,which can be captured at atomic resolution with MD simulations [192,193].The combination with protein dynamics pushes the boundaries of de novo design and emphasizes the importance of accurate modeling of the local energy landscape for designing protein dynamics.The predictive power of computational design has stimulated the discovery of industrial enzymes,enabling the synthesis of new biomaterials.
4.Perspectives
As mentioned above,automatic retrobiosynthesis and computational enzyme design have developed rapidly in the last decade.The establishment of new methods and the development of new tools have entered the age of blooming flowers.Among them,retrobiosynthesis is gradually transforming from a simple auxiliary tool to a full extension of the scientist’s brain with the help of artificial intelligence technology.The de novo enzyme design has realized the guidance of researchers to complete the engineering transformation of molecular production lines at the atomic level.The picture of mankind fully grasps nature’s “production technology” has appeared in thevisible range in the future.But seeing does not mean achieving,the integration of the two technologies still lacks effective methods and tools.The pathway designed by retrobiosynthesis is often unreasonable,and computational enzyme design often hard to find most suitable design goal.The close complementarity between the two has not yet been formed.
However,the foundation of integration is gradually being established.The work of Siegel et al.successfully demonstrated how a hypothetical biosynthetic pathway can function with the aid of computational enzyme design.Although the design of the pathway itself still relies on the experience of scientists rather than computer assistance,it is enough to allow researchers to see the great value brought by the complementarity of the two technologies.In addition,databases of known enzyme and their corresponding detailed catalytic mechanisms have also begun to appear.These databases are expected to rely on the name of enzymes to be linked to retrobiosynthesis,while relying on catalytic mechanisms to correlate to computational enzyme design.In short,it can be expected that in the not-too-distant future,computers will be able to help the industry design any desired biomanufacturing process plan for target chemicals,and an intelligent biomanufacturing era will come.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
We gratefully acknowledge the support from the National Natural Science Foundation of China (U1663227,21861132017,21811530003,21878170).
杂志排行

Chinese Journal of Chemical Engineering的其它文章
- Struggling as in past, write a glorious future together-CJChE’s 40th anniversary
- Reduced graphene oxide modified melamine sponges filling withparaffin for efficient solar-thermal conversion and heat management
- Photocatalytic degradation of tetracycline hydrochloride with visible light-responsive bismuth tungstate/conjugated microporous polymer
- Ag nanoparticles anchored on MIL-100/nickel foam nanosheets as an electrocatalyst for efficient oxygen evolution reaction performance
- Performance improvement of ultra-low Pt proton exchange membrane fuel cell by catalyst layer structure optimization
- Anodic process of stibnite in slurry electrolysis:The direct collision oxidation