陆地棉吐絮率的限制性两阶段多位点全基因组关联分析及候选基因预测
2022-02-24谢晓宇王凯鸿秦晓晓王彩香史春辉宁新柱杨永林秦江鸿李朝周马麒宿俊吉
谢晓宇,王凯鸿,秦晓晓,王彩香✉,史春辉,宁新柱,杨永林,秦江鸿,李朝周,马麒✉,宿俊吉✉
1甘肃农业大学生命科学技术学院/省部共建干旱生境作物学国家重点实验室,兰州 730070;2新疆农垦科学院棉花研究所,新疆石河子 832000;3石河子农业科学研究院,新疆石河子 832000
0 引言
【研究意义】棉花(Gossypium spp.)是一种生产天然纤维的重要经济作物,在中国国民经济中占有重要地位。西北内陆是中国棉花的主产区,但该地区有效积温相对较低、无霜期相对较短,更适于早熟棉花品种的种植。因此,在西北内陆棉区,棉花的早熟性显得至关重要。吐絮率是指吐絮周期内某个特定时期吐絮棉铃数占总棉铃数的比率,是衡量棉花吐絮集中性的重要指标,也是棉花早熟性的一个重要性状。因此,深入解析陆地棉吐絮率的遗传基础,鉴定其QTL等位基因,以及挖掘与目标性状相关的候选基因,对于培育早熟和适宜机采棉花新品种等方面具有重要意义。【前人研究进展】棉花早熟性是一个综合性状,其相关性状主要包括霜前花率、果枝始节位、果枝始节高、现蕾期、开花期、花铃期和全生育期等[1-6]。棉花早熟性是受多基因控制的数量性状,存在加性和显性效应[7]。相对于产量和纤维品质性状,有关棉花早熟性状的研究相对较少。人们利用连锁分析的方法,采用简单重复序列(simple sequence repeats,SSR)标记和单核苷酸多态性(single nucleotide polymorphism,SNP)标记定位到棉花全生育期、开花期等早熟相关性状的数量性状位点(quantitative trait locus,QTL)[8-11]。随着高通量测序技术的快速发展,全基因组关联分析(genome-wide association studies,GWAS)已成为检测QTL的另一种重要方法,以遗传变异更丰富的自然群体为研究材料,基于连锁不平衡的关联作图方法,大大地提高了QTL的检测效率[12]。GWAS已被广泛用于水稻[13]、玉米[14]、油菜[15]、大豆[16]和棉花[17-18]等作物育种目标性状定位的研究中。目前,已报道棉花GWAS研究主要集中在纤维品质和产量等性状上[19-23],关于早熟性GWAS的研究相对较少[18,24-25]。SU等[24]利用简化基因组测序开发的SNP标记,开展了6个早熟相关性状的GWAS研究,鉴定出8个SNP位点与5个早熟相关性状之间存在显著关联,在与多个早熟相关性状同时显著关联的1个SNP标记附近发现一个调控开花的候选基因 CotAD_01947。LI等[25]利用 80K SNP基因芯片对169份陆地棉的早熟性状进行了大量的研究,获得29个显著的SNP位点,确定了2个潜在的改良棉花早熟性的候选基因 Gh_D01G0340和Gh_D01G0341。LI等[18]利用基于全基因组重测序GWAS鉴定到了与7个早熟相关性状显著关联的307个SNP位点及1个候选基因Ghir_D03G011310。近年来,随着劳动力资源的紧缺和棉花人工采摘成本的大幅上涨,机械采收已成为西北内陆棉花收获的主要方式,而机械采收前需要对棉花进行脱叶催熟处理。然而,脱叶催熟技术对棉花产量和纤维品质有一定的影响。为了减小化学脱叶对棉花产量和纤维品质的影响,西北内陆棉区生产上一般要求9月中旬棉田整体吐絮率达到50%以上,才能喷施脱叶催熟剂[26-27],因此,9月中旬棉花吐絮率已成为评价品种是否适宜机械采收的关键性状之一。然而,目前对陆地棉吐絮率的遗传解析及QTL定位的研究很少。李俊文等[28]基于连锁作图方法,利用陆地棉重组近交系(recombinant inbred lines,RIL)和永久 F2群体,对控制陆地棉吐絮性状进行 QTL检测,共检测到了 11个与吐絮率相关的QTL。目前,GWAS方法已被广泛应用于农作物育种目标性状的遗传解析,但是有关利用GWAS挖掘陆地棉吐絮率QTL的研究鲜有报道。而且GWAS的混合线性模型(mixed linear model,MLM)不能检测自然群体中广泛存在的复等位变异。同时进行多重测验矫正时,通常利用提高显著水平使得该模型仅能检测到少数关联位点,无法较为全面地检测到QTL位点。针对目前常规MLM-GWAS方法的局限性,HE等[29]研究提出了一种限制性两阶段多位点全基因组关联分析(restricted two-stage multi-locus GWAS,RTM-GWAS)方法,该方法是一种新发现的可以全面检测自然群体和双(多)亲衍生群体中具有不同复等位变异QTL的分析方法,它解决了常规GWAS方法不能估计复等位变异、不能充分检测出全基因组QTL等问题,大大提高了QTL的检测效率[30]。RTM-GWAS方法已成功应用于大豆百粒重[31-32]、油酸含量[33]、蛋白质含量[34]和异黄酮含量等[35],棉花产量[36]、纤维品质[23]等数量性状遗传基础的解析。【本研究切入点】目前,西北棉区喷施脱叶剂是棉花机械采收的重要环节之一,且吐絮率已成为喷施脱叶剂之前一个很重要的评价指标。因此,解析吐絮率性状的遗传基础,对于快速培育机采棉专用品种方面具有重要意义。然而,目前有关陆地棉吐絮率性状遗传基础解析的研究很少,使得吐絮率的研究落后于其他早熟相关性状。【拟解决的关键问题】本研究采用RTM-GWAS方法对315份陆地棉材料的吐絮率性状展开研究,挖掘控制吐絮率性状的优异等位变异位点,并进行候选基因预测,以期解析陆地棉吐絮率的遗传基础,为陆地棉早熟和适宜机采相关性状的分子育种奠定理论基础。
1 材料与方法
1.1 试验材料与田间试验
甘肃农业大学棉花分子育种实验室前期构建了由315份陆地棉种质材料构成的一个自然群体,包含290份来自中国不同时期、不同地域选育的品种(系),25份从国外引进的品种(Foreign Region,FR)[36]。290份国内材料按照地域来源进行分类,125份来源与黄河流域棉区(Yellow River Region,YRR)、48份来源于长江流域棉区(Yangtze River Region,YZRR)、97份来源于西北内陆棉区(Northwest Inland Region,NIR)和20份来源于北部特早熟棉区(Northern Super Early-maturing Region,NSER)(电子附表 1)。2014—2015年,将该陆地棉自然群体的315份材料种植于中国农业科学院棉花研究所安阳老所部试验田(河南安阳,2个环境分别命名为AY-14和AY-15);2014年种植于新疆农垦科学院棉花研究所试验田(新疆石河子,该环境命名为SHZ-14)。每个环境设置3个重复,采取随机区组试验设计方案,田间管理参照当地常规方法。河南安阳试验点:每个材料单行种植,行长5.00 m,行距0.80 m,株距0.20 m,每个材料约23个单株;新疆石河子试验点:每个材料分2行种植,行长2.00 m,行距0.45 m,株距0.10 m,每个材料约40个单株。AY-2014和AY-2015均于当年的4月19日播种,SHZ-2014于4月25日播种。
1.2 表型性状鉴定与数据分析
在各试验点的材料正常吐絮后,对每个材料的每个重复,挑选长势整齐、均匀一致的10株单株进行系红线标记。3个环境(AY-14、AY-15和SHZ-14)的吐絮率调查时间分别为9月18日、9月10日、9月28日,调查上述被标记10个单株上的总棉铃数及吐絮棉铃数(单株中直径大于2.0 cm的棉铃被定为有效棉铃)。计算每个种质材料的吐絮率,其计算公式为:吐絮率=(吐絮棉铃数/总棉铃数)×100%。然后利用SPSS 26.0软件对获得吐絮率的表型数据进行统计描述和方差分析,并计算其广义遗传力。
1.3 全基因组关联分析(GWAS)
陆地棉自然群体的 SNPLDB标记构建来源于甘肃农业大学棉花分子育种实验室前期的研究基础[36]。利用SLAF-seq(specific-locus simplified fragment sequencing)方法对自然群体进行SNP位点基因分型,参考陆地棉TM-1 v2.1基因组[37]对测序所获得的序列进行比对,然后根据最大缺失率>0.10,最小等位基因频率(minor allele frequency,MAF)<0.05的标准过滤挖掘,获得13 391个高质量SNP位点。将分布在同一连锁不平衡(linkage disequilibrium,LD)区块内的多个紧密连锁SNP标记,记为同一个SNP连锁不平衡区段(SNP linkage disequilibrium block,SNPLDB),获得9 244个SNPLDB标记[36]。然后利用这些SNPLDB标记和3个种植环境下吐絮率的表型值,对 AY-14、AY-15、SHZ-14 3个单环境表型和多环境联合分析获得的表型分别进行RTM-GWAS分析。利用SAS软件PROC GLM 程序中方差分析的线性模型,按照郝晓帅等[38]提出的方法,估算获得多环境联合分析的表型值。然后按照HE等[29]提出的RTM-GWAS程序进行关联分析,该程序主要分为2个分析阶段:(1)基于单位点简单线性模型对SNPLDB标记进行初步筛选;(2)使用多位点逐步回归模型进行筛选,同时估计每个显著位点的等位基因效应值。根据等位基因效应值,利用 R 3.6.0软件建立目标性状的QTL-allele矩阵。在RTMGWAS 2个阶段的分析程序中,显著水平(P)均设置为0.05。另外,利用R 3.6.0软件,以PCs和亲缘关系系数K为协变量进行MLM-GWAS分析,鉴定吐絮率性状的显著SNP位点,利用R 3.6.0软件绘制出关联位点的曼哈顿图,以-lg(P)≥4.43的SNP为显著位点。
1.4 稳定关联位点优异等位变异的鉴定
能在多环境联合分析、单环境2个分析模式中同时检测到的SNPLDB位点,被认为与吐絮率显著关联的稳定关联位点。利用 SPSS 26.0软件计算稳定SNPLDB位点各种等位变异类型表型的平均值,并进行双尾T检验,确定优异等位变异类型,使用Origin 2018绘制相对应的统计图。
1.5 优异等位变异类型频率的比较分析
在315份陆地棉种质材料中,分别选取拥有最高和最低吐絮率的品种系各50个,分析比较稳定关联位点优异等位变异类型的频率;然后比较YRR、YZRR、NIR和NSER 4个不同地理来源种质材料间优异等位变异类型的频率差异。
1.6 候选基因的挖掘
根据SNPLDB位点在陆地棉TM-1 v2.1参考基因组[38]上的物理位置,结合 LD的衰减距离,将稳定显著关联SNPLDB位点的两端各扩展500 kb,形成约1 Mb基因组区间。利用棉花功能基因组学数据库(https://cottonfgd.org/),列出分布在上述显著关联的稳定SNPLDB位点两侧1 Mb区间内的全部基因。然后分别利用南京农业大学(Nanjing Agricultural University,NAU)和中国农业科学院棉花研究所(Cotton Research Institute of Chinese Academy of Agricultural Sciences,CRI)2组不同组织的RNA-Seq数据,计算它们的FPKM(fragments per kilobase of transcript per million fragments mapped)值,筛选出在棉铃胚珠和纤维组织中高表达的基因作为候选基因,并对候选基因进行功能注释。候选基因表达方式的热图(heatmap)和维恩图(venn diagram)分析使用OmicShare在线工具(http://www.omicshare.com/tools)绘制完成。最后利用棉花功能基因组学数据库对候选基因进行GO富集分析和KEGG代谢途径分析。
2 结果
2.1 陆地棉吐絮率的表型变异特征
315个陆地棉品种(系)吐絮率在3个环境中的变异范围为 39.27%—91.32%,平均值为 69.14%,变异系数为14.25%。AY-14环境下的吐絮率分布范围相对较小,变幅为46.73%—90.00%,平均值为80.62%,变异系数为9.86%。AY-15环境下的吐絮率分布范围相对较大,变幅为 10.42%—100.00%,平均值为55.99%,变异系数为34.60%。SHZ-14环境下的吐絮率分布范围也较小,变幅为37.78%—98.05%,平均值为70.80%,变异系数为14.42%(表1)。3个环境下吐絮率的方差分析结果显示,基因型、环境以及基因型×环境互作均呈现极显著差异(P<0.0001),说明吐絮率的基因型与环境之间存在显著的互作效应(表2)。利用3个环境计算的吐絮率广义遗传力相对较大,为67.03%(表2)。以上结果表明,陆地棉吐絮率性状主要受遗传因素影响,也受环境因素影响。
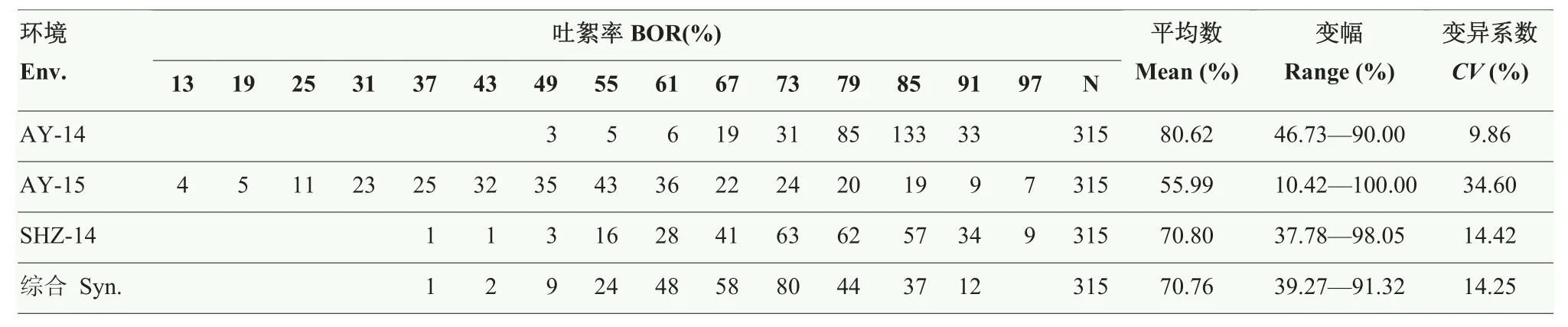
表1 陆地棉吐絮率的次数分布和描述统计Table 1 Frequency distribution and descriptive statistics of boll opening rate in upland cotton
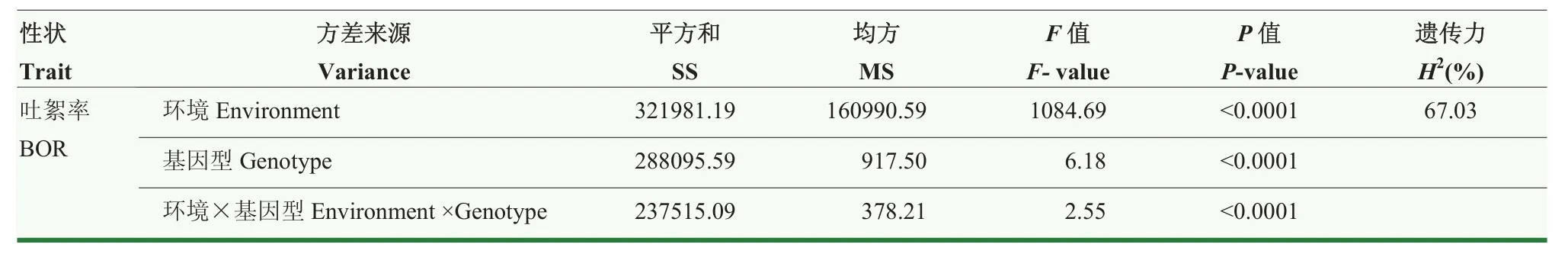
表2 陆地棉吐絮率的方差分析Table 2 Variance analysis of boll opening rate in upland cotton
2.2 全基因组关联分析(GWAS)
甘肃农业大学棉花分子育种实验室前期已利用SNPLDB标记对陆地棉自然群体进行了聚类分析和主成分分析(principal component analysis,PCA),把315个品种(系)划分为2个类群;LD分析发现该群体在全基因组水平上的平均LD值约为500 kb[36]。通过RTM-GWAS方法的多环境分析模型,在9 244个SNPLDB中检测到52个SNPLDB位点与吐絮率显著关联,这些显著关联位点分布在陆地棉全部26条染色体上,每条染色体上分布1—5个位点(表3)。第2、4、6、7、14、18、20、24和26染色体上最少,仅检测到1个SNPLDB标记;第15染色体上最多,检测到5个SNPLDB标记。在52个显著关联的SNPLDB位点中,10个关联位点的-lg(P)值大于10.00,其中,LDB_16_37952328上具有最高的-lg(P)值(46.14)。进一步分析发现,在多环境联合分析中检测到的 52个显著关联位点中,8个位点同时也至少在1个单环境中也被检测到,例如,位点LDB_16_37952328在2个单环境(AY-15和SHZ-14)中被检测到;LDB_5_96395565、LDB_16_49503485、LDB_10_6908012、LDB_15_9697358和LDB_4_81118668在AY-15单个环境中被检测到。根据显著关联位点的-lg(P)≥10,以及在单环境和多环境联合分析中同时出现的稳定性,最终筛选出6个SNPLDB可能是与吐絮率稳定显著关联的主效位点,它们分别为 LDB_16_37952328、LDB_5_96395565、LDB_16_49503485、LDB_10_6908012、LDB_15_9697358和LDB_4_81118668(图1-A)。这6个位点均为单个SNP构成SNPLDB位点,共可解释7.04%的表型变异(表3)。本研究利用MLM-GWAS方法仅发现1个位点SNP_16_37952328与吐絮率显著关联(图1-B)。
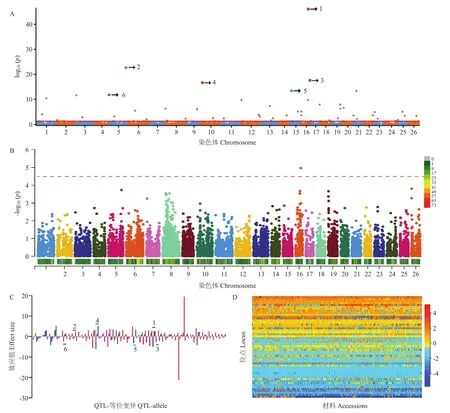
图1 利用RTM-GWAS和MLM-GWAS方法对陆地棉吐絮率的遗传解析Fig.1 Genetic analysis of boll opening rate in upland cotton by MLM-GWAS and RTM-GWAS procedure

表3 与陆地棉吐絮率显著关联的SNPLDB位点Table 3 SNPLDB loci significantly associated with boll opening rate in upland cotton
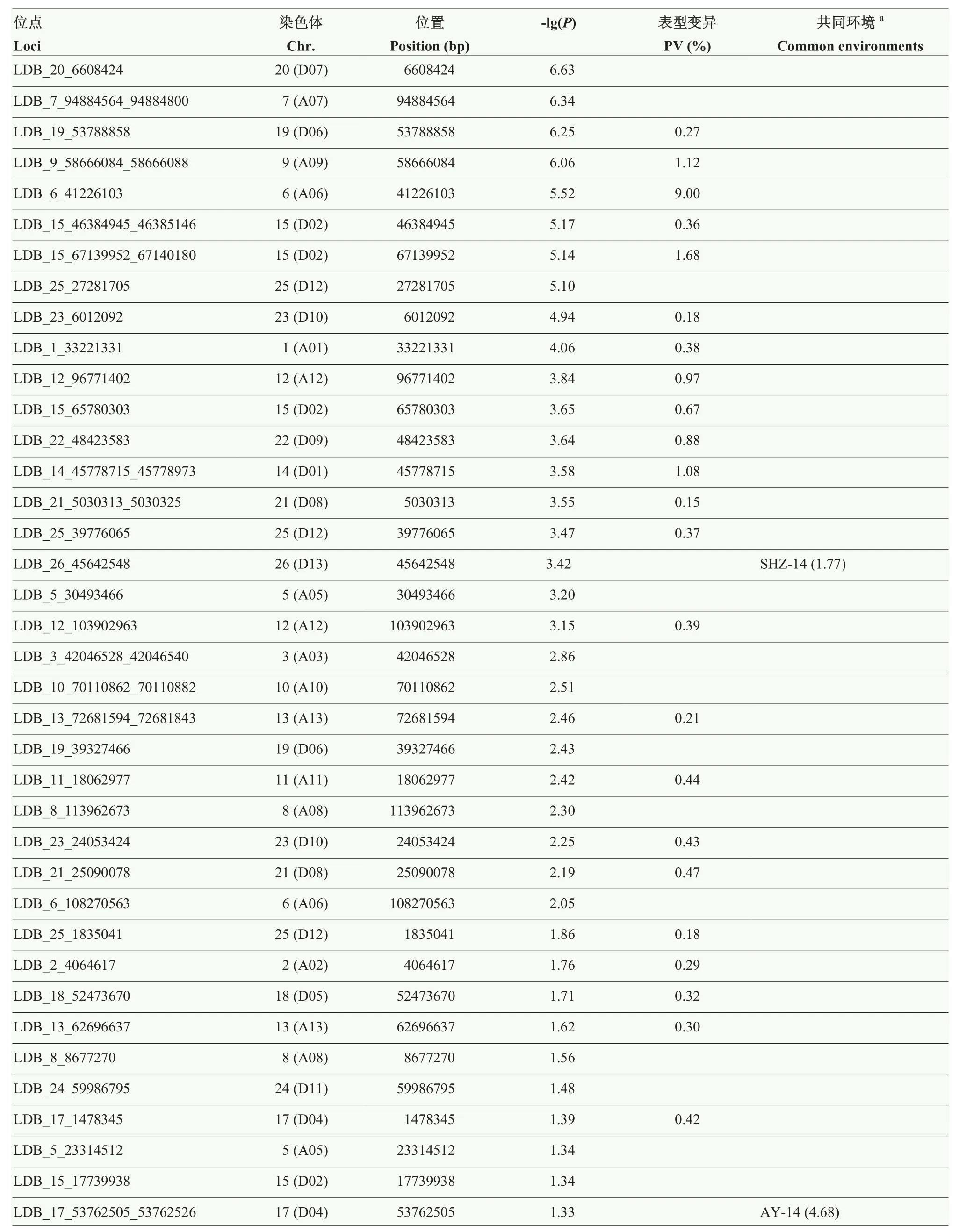
续表3 Continued table 3
52个显著关联的SNPLDB位点中,38个为单个SNP的等位变异位点,14个为含有多个SNP的复等位变异位点。这些显著关联SNPLDB位点等位变异类型的分布范围为3—10个,共179个,其中90个为增效等位变异/单倍型,89个为减效等位变异/单倍型。增效等位变异类型的效应值分布在0.014—19.43,减效等位变异类型的效应值分布在-21.49—-0.039。显著关联位点中,复合位点LDB_19_52309050_52309284的等位变异效应值变异范围较大(-21.49—19.43),其他等位变异/单倍型的效应值变异范围较为集中,约96%的等位变异/单倍型效应值分布在-6—5(图1-C)。为了更加明晰地展示陆地棉种质群体的遗传基础,利用52个显著SNPLDB位点的等位变异效应值,构建了一个 52×315(位点×品种系)的QTL等位变异矩阵,该矩阵能够直观地反映了315个陆地棉种质系中控制吐絮率的遗传变异构成(图1-D)。
2.3 稳定显著关联位点优异等位变异的鉴定
为了鉴定上述6个稳定显著关联位点的优异等位变异类型,对其每种等位变异所对应的吐絮率进行了显著性比较。关联位点LDB_16_37952328存在有CC(n=217)、CT(n=23)和TT(n=67)3种等位变异类型,携带有TT和CT等位变异种质的吐絮率(分别为80.08%和77.17%)极显著高于携带有CC的种质系(67.07%)(图2-A)。LDB_5_96395565存在AA(n=212)、AG(n=17)和 GG(n=53)3种等位变异类型,AA型的吐絮率(71.87%)极显著高于 GG型(67.26%)(图2-B)。LDB_16_49503485存在有CC(n=36)、CT(n=30)和TT(n=249)3种等位变异类型,TT型种质的吐絮率平均值(71.12%)显著高于含有CC型(67.04%)和CT型(67.25%)(图2-C)。LDB_4_81118668存在 CC(n=76)、CT(n=37)和TT(n=182)3种等位变异类型,CT型(72.84%)和 TT型(71.82%)种质系的吐絮率极显著高于 CC型(67.83%)(图 2-D)。其余 2个稳定关联位点LDB_10_6908012和LDB_15_9697358的3种等位变异类型所对应种质的吐絮率无显著性差异(图2-E和图 2-F)。按照不同等位变异类型对应种质的吐絮率有显著性差异,吐絮率高的等位变异类型为优异等位变异原则,4个稳定显著关联位点及其优异等位变异被鉴定,其优异等位变异分别为 LDB_16_37952328(TT)、LDB_5_96395565(AA)、LDB_16_49503485(TT)、和LDB_4_81118668(TT)。

图2 6个稳定显著关联SNPLDB位点不同等位变异的吐絮率比较Fig.2 Comparison of boll opening rate with different allelic variation at six stable and significant association SNPLDB loci
2.4 优异等位变异分布频率的比较
为了研究优异等位变异分布频率与吐絮率的关系,分别选取2组高、低吐絮率的极端材料各50份。高吐絮率种质的表型分布在81.88%—91.32%,低吐絮率种质的表型分布在 39.27%—60.59%。比较结果发现,4个稳定关联SNPLDB位点在高吐絮率品种(系)中的优异等位变异频率均高于低吐絮率品种(系)(图3-A)。
为了比较不同地理来源的材料间吐絮率及其优异等位变异频率的差异性,选取来自YRR、YZRR、NSER和NIR4个不同地理来源区域的125、48、20和95个品种(系)。不同地理来源品种(系)的吐絮率平均值大小顺序为NSER(75.91%)>YRR(73.95%)>NIR(69.96%)>YZRR(64.72%)。方差分析结果表明,NSER品种(系)的吐絮率极显著高于 NIR和YZRR,且YRR和NIR品种(系)的吐絮率极显著高于YZRR(P<0.01,图3-B)。此外,对4个稳定关联位点的优异等位基因频率进行比较,发现4个位点的优异等位基因频率在中国 4个不同地理来源区域的品种(系)间存在差异。进一步分析发现,位点LDB_16_49503485在YRR、YZRR和NIR有较高的优异等位基因频率,均在 70%以上(图3-C)。另外3个稳定关联位点(LDB_16_37952328、LDB_5_96395565和LDB_4_81118668),在YRR和NSER品种(系)中的优异等位变异位点频率要高于来源于YZRR和NIR的品种(系)(图3-D—F),优异等位变异频率的变化与不同地理来源品种(系)的吐絮率表型性状变化趋势一致。
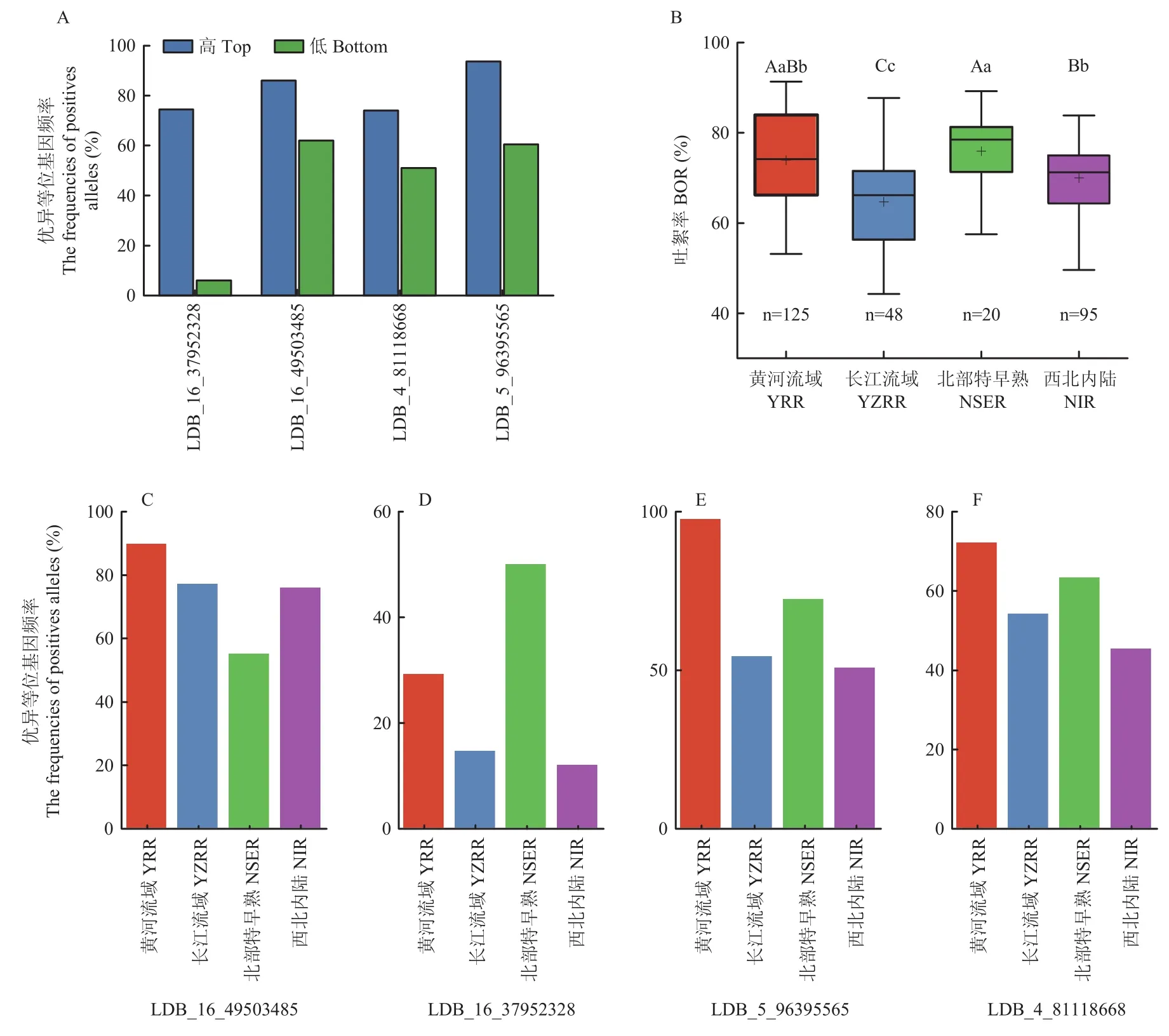
图3 4个稳定关联位点优异等位变异的频率分布Fig.3 Frequency distribution of superior alleles of four stable association loci
2.5 候选基因的预测
根据陆地棉TM-1v2.1参考基因组[38],在4个稳定关联位点两侧1 Mb扩展区域中存在178个基因。利用NAU测序获得的RNA-seq计算基因FPKM值,发现其中61个基因,至少在陆地棉10个组织或器官中一个中有较高表达量。依据基因在不同组织中的表达情况,可把61个基因分为4组(用1—4表示)。第1组(17个)主要在第5、10和20 DPA(days post anthesis,开花后生长天数)的纤维中高表达;第2组(9个)主要在花瓣和雄蕊中高表达;第3组(17个)主要在胚珠的8个发育阶段中高表达;第4组(18个)主要在根、茎、叶组织和花器官中较高表达(图4-A)。利用另一组CRI测序获得的RNA-seq数据,发现59个基因至少在 12个组织或器官的一个中有较高表达量,将59个基因分为4组(用I—Ⅳ表示)。第Ⅰ组(16个)在花瓣、花托、花萼、苞片、花药和花丝中高表达;第Ⅱ组(13个)在第10、15、20和25 DPA的纤维中高表达;第Ⅲ组(9个)在根、茎和叶中高表达;第Ⅳ组(21个)在第10、15、20和25 DPA的胚珠中高表达(图4-B)。
通过NAU和CRI的2组RNA-seq数据,各发现34个基因在胚珠和纤维中高表达。它们中的23个基因在两组数据中的表达模式一致,推测它们可能是与陆地棉吐絮率相关的候选基因(图 4-C)。将预测的23个候选基因可分为3类基因,a类基因(9个)主要在0—5 DPA的胚珠中表达,b类基因(4个)主要在10—35 DPA的胚珠中表达,c类基因(10个)主要在5—25 DPA的纤维中表达。进一步分析发现,20个候选基因具有生物学功能注释,其中过氧化物酶体(S)-2-羟基氧化酶基因Gh_D03G1603和钙结合线粒体载体蛋白基因 Gh_D03G1058可能是促进棉铃成熟或早衰的调控基因(表4)。

表4 与陆地棉吐絮率相关的候选基因Table 4 Candidate genes related to BOR in upland cotton
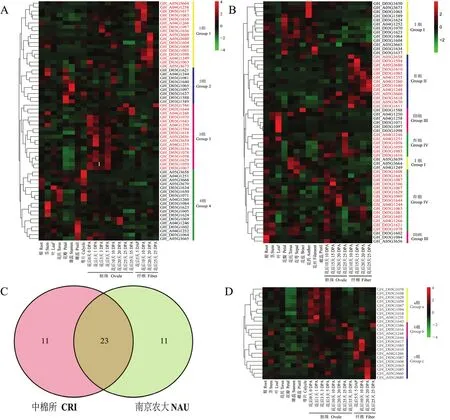
图4 与陆地棉吐絮率相关候选基因的预测Fig.4 Prediction of candidate genes related to the BOR of upland cotton
对上述预测的23个候选基因进行GO富集分析和KEGG代谢途径分析。GO结果显示,在20个基因中总共发现了 55个调控途径,其中 Gh_D03G1608、Gh_D03G1610、Gh_D03G1616、Gh_D03G1629和Gh_D03G1067与蛋白质氨基酸糖蛋白结合相关(GO:0005515)。KEGG结果显示,在11个基因中发现了32个调控途径,其中 Gh_A05G3660、Gh_D03G1063和Gh_D03G1067有共同的核糖体代谢途径(ko01100)(电子附表2)。
3 讨论
3.1 吐絮率在机采棉生产中的重要性
2020年全国棉花播种面积已达316.99万hm2,总产量为591.00万t;西北内陆区棉花种植面积为251.85万hm2,总产量为519.10万t,其产量占全国棉花总产量的 87.83%(http://www.stats.gov.cn/tjsj/zxfb/202012/t20201218_1810113.html)。棉花已成为西北内陆最主要的经济作物之一。然而该地区春季温度回升慢、秋季下降快、初霜来临早,一旦发生霜冻,会造成棉花减产,因此,培育早熟棉品种显得尤为重要[39]。早熟性也是适宜机采棉品种最为关键的性状之一。机械采棉是目前降低中国棉花生产成本,实现棉花生产可持续发展,提升棉花国际竞争力的重要措施之一。在西北内陆棉区,为了提高棉花机收效率而不降低其产量和纤维品质,所以在9月中下旬棉田整体吐絮率达到50%以上时,才喷施脱叶催熟剂。如果棉田吐絮率低于50%时,喷施脱叶催熟剂会显著降低中上部棉铃的单铃重及纤维品质,从而造成棉花减产降质,因此,吐絮率已成为西北机采棉育种中不可忽视的一个重要性状。
棉花从播种出苗到群体一半的第一个棉铃吐絮所需的天数称为全生育期。该性状全生育期因品种、气候及栽培条件的不同而异,通常陆地棉品种全生育期约为100—140 d。然而全生育期并不包含吐絮期(第一棉铃吐絮到最后一个棉铃吐絮的时间段)的生育阶段。以前的研究中,把霜前花率作为评价吐絮期吐絮进程的一项重要指标[1-2]。霜前花率是指早霜以前及早霜后5 d内所采摘的棉花产量占总产量的百分率。然而该指标在目前的机采棉生产模式下已失去了实际意义,无法指导当前的机采棉生产。如果一个棉花品种的吐絮期过长,造成吐絮不够集中,会影响机械化采收进程,故吐絮集中已成为早熟的一个关键性状。吐絮率不仅仅是一个反映棉花早熟性的重要指标之一,而且是反映吐絮集中程度的重要指标。吐絮越早越集中,吐絮率越高,反之则越低。然而目前有关棉花吐絮率的研究主要集中在脱叶剂方面[40],缺乏从遗传基础方面的解析和研究,对控制棉花吐絮率QTL定位和候选基因挖掘等方面的研究较少。本研究中全面解析陆地棉吐絮率性状遗传基础,挖掘调控棉花吐絮率QTL及候选基因,对于今后开展早熟机采棉分子育种具有重要意义。
3.2 与吐絮率稳定关联位点及其优异等位变异的发掘
准确的表型鉴定是利用GWAS挖掘农作物目标性状优异等位变异和候选基因的关键。本研究发现315个陆地棉品种(系)吐絮率存在广泛的表型差异,且3个环境间表型值存在极显著差异。其原因是吐絮率大小易受温度、光照、有效积温等外界环境因素的影响。在不同环境种植陆地棉种质材料,由于鉴定吐絮率的外界环境因素在不同地点、不同年份之间会存在不小的差异,因此获得表型性状也会存在较大的差异。如AY-2014和AY-2015,同一地点不同年份之间其表型数据也存在显著差异。故分别利用单个环境的表型值和多环境联合估算的表型值进行了 RTM-GWAS分析,鉴定能够与目标性状稳定关联SNPLDB位点。本研究中利用多环境联合估算的表型值进行了 RTM-GWAS分析,共获得了52个SNPLDB位点与吐絮率显著关联。将该52个与吐絮率显著关联的SNPLDB位点与前人发现的其他早熟性状相关 QTL进行比较,发现位点LDB_16_37952328、LDB_17_51465593、LDB_5_23314512和 LDB_21_22036171与前人研究报道的QTL相重叠[10,25,41-42]。其中分布在D03染色体上位点LDB_16_37952328在多篇文章中被发现[23-24,28],这些结果证实了本研究结果的可靠性,也证明该位点是控制陆地棉早熟相关性状的一个关键位点。它不仅与开花期和全生育期显著相关,而且与吐絮率显著相关,值得在后期的研究中重点关注。此外,其余48个位点应该是新发现与吐絮率相关的位点。总之,目前与其他早熟相关性状 QTL或关联位点的研究相对较多[3,5,22,28],但有关与棉花吐絮率稳定关联的位点或候选基因的研究很少。本研究利用两组前人报道的 RNA-seq数据,发现 23个基因在胚珠和纤维中高表达,推测它们可能是与陆地棉吐絮率相关的候选基因。更为重要的是,推测Gh_D03G1603和Gh_D03G1058可能通过调节棉铃成熟或早衰,促进棉花集中早絮。Gh_D03G1603是过氧化物酶体(S)-2-羟基氧化酶基因(GLO1),水稻中该类基因通过改变H2O2含量来调节生长素含量,进而促进水稻早熟[43]。由此我们推测 Gh_D03G1603通过调控棉花子房中的生长素含量提高,促进子房及其周围组织的膨大,加速了棉铃发育;当生长素达到一定含量时还会诱导乙烯的形成,进而促进棉花集中早絮成熟。Gh_D03G1058是钙结合线粒体载体蛋白基因(slc25a24),拟南芥中调节激活Ca2+运输通道介导脱落酸合成,导致气孔关闭,促进拟南芥早衰[44]。推测Gh_D03G1058可能调节棉铃中Ca2+运输,激活大量脱落酸合成,促进棉铃的发育和成熟。
根据显著关联位点的-lg(P)值的大小以及在多环境联合分析和单环境分析中显著位点同时出现的稳定性,在52个SNPLDB位点中最终筛选出6个位点可能与吐絮率稳定关联的主效位点。通过不同等位变异类型对应种质的吐絮率的显著差异比较分析,最终确定4个位点是稳定显著关联的主效位点,并鉴定出该4个主效位点的优异等位变异类型。进一步分析发现,4个稳定关联主效位点的优异等位变异在高吐絮率种质系中出现频率高于低吐絮率种质系中出现的频率。例如,特早熟陆地棉优良品种新石 K18中,同时包含了该4个与吐絮率关联SNPLDB位点的优异等位变异类型。此外,在4个不同地理来源的品种系间,YRR和NSER的平均吐絮率较高,YZRR和NIR的平均吐絮率较低,同时位点LDB_16_37952328、LDB_5_96395565、和LDB_4_81118668在4个不同地域的优异等位变异频率与之相对应,进一步确定了这些优异等位变异的可靠性。因此,可以根据这些优异等位变异筛选出优良种质材料,为分子标记辅助选择提供优异亲本资源。
3.3 RTM-GWAS方法的优越性
RTM-GWAS方法能够较全面地解析陆地棉吐絮率性状QTL及其复等位变异,分析结果用于个别基因挖掘的同时,还可用于群体遗传结构分析以及作物育种的优化组合设计等方面[30]。RTM-GWAS方法基于包含不同单倍型/等位基因的 SNP连锁不平衡区块,这种方法比常规GWAS更适合QTL鉴定;该方法可以提供具有比较总体遗传信息的QTL-等位基因矩阵,包括显著的单核苷酸多态性位点及其等位基因效应、群体等位基因组成和每个位点等位基因频率的差异,这是以往传统的单位点GWAS模型无法做到的[45-46]。本研究利用RTM-GWAS方法对陆地棉吐絮率性状进行分析,检测到52个显著关联SNPLDB位点,其中4个位点在多环境联合分析和单环境分析中同时被检测到,被认定为稳定关联的主效SNPLDB位点;而利用MLM-GWAS进行吐絮率的研究时,仅检测到1个显著 SNP位点。此外我们在前期利用 MLM-GWAS进行其他6个早熟相关性状的研究中,检测到8个SNP位点与5个早熟性状之间存在13个显著关联,发现1个稳定SNP位点(rsDt3:13562854)同时多个早熟相关性状(-lg(P)≥6.21)之间存在显著关联[24]。这些结果都充分证明了RTM-GWAS方法在全基因组关联位点检测方面具有优势。
4 结论
在陆地棉自然群体中检测到52个与吐絮率性状显著相关的SNPLDB位点。其中4个被鉴定为与吐絮率稳定关联的主效SNPLDB位点。在该4个主效位点的邻近区域共注释了178个基因,预测发现了其中的 23个基因可能是调控陆地棉吐絮率的候选基因。
