江西省2020年新冠肺炎病例临床样本SARS-CoV-2全基因组测序及基因特征研究
2022-02-23李健雄施勇刘师文徐刚龚甜周珺肖芳刘晓庆张艳妮肖大瑾冉鑫熊英
李健雄,施勇,刘师文,徐刚,龚甜,周珺,肖芳,刘晓庆,张艳妮,肖大瑾,冉鑫,熊英
(江西省疾病预防控制中心,江西 南昌 330029)
2020 年初,新型冠状病毒肺炎(Corona Virus Disease 2019,COVID-19)[1](简称” 新冠肺炎” )迅速蔓延,席卷全世界,对全球的经济、社会造成了巨大的冲击。目前,全球确诊病例超1.8亿例,死亡破400万例,新冠肺炎疫情防控形式十分严峻。
SARS-CoV-2为线性单股正链的RNA病毒,属于β属的冠状病毒,有包膜。其基因组长约为29.8 Kb,基因特征与SARS-CoV和MERS-CoV有明显区别[2]。COVID-19患者和无症状感染者均可能通过呼吸道飞沫、密切接触等途径将SARSCoV-2传播给易感人群[3]。由于病毒的复制过程易出错特性,特别是RNA病毒,随着时间的推移积累突变将导致序列的改变多样性。文献报道[4],目前SARS-CoV-2的核苷酸突变率估计约为2.5个核苷酸/月,其基因组变异可能会对其传播力、致病力产生影响。因此,快速、准确的进行SARS-CoV-2基因组测序,掌握基因组特征,对疫情防控具有非常重要意义。
2020年1 至3月,江西省共发现930例新冠肺炎确证病例,是除湖北省以外疫情较为严重的几个省份之一。后期,在外防输入阶段,我省共发现5例境外输入病例。为了了解江西省病毒基因变异情况,建立江西省本土新冠病毒基因库,为后续新冠疫情的溯源提供参考依据,本研究于2020年1月、2020年2月和2020年9月,分别建立了新冠肺炎病例临床样本的二代和三代基因组测序技术,并应用于新冠肺炎确证病例临床样本的SARS-CoV-2全基因组测序,现将研究结果报道如下。
1 材料与方法
1.1 样本来源 样本来源为2020年江西省新冠肺炎确诊病例的咽拭子和痰液等呼吸道样本。
1.2 核酸提取 采用Qiagen RNeasy Mini Kit(Qiagen,Cat No:74104)对38份新冠肺炎病例的咽拭子和痰液等呼吸道临床样本进行病毒核酸提取,具体操作步骤参照试剂盒说明书。
1.3 文库构建及全基因组测序
1.3.1 基于Ion Torrent S5平台的二代宏基因组测序 建立基于Ion Torrent S5平台的新冠二代宏基因组测序方法,采用Nugen OvationRNA-Seq System、Ion XpressTM Plus Fragment kit或 者Ion total RNA-Seq Kit等试剂盒构建测序文库。采用Ion OneTouch2系统构建测序模板。使用Thermofisher测序平台的Ion Torrent S5测序仪和Ion530芯片进行全基因组深度测序。以SARSCoV-2 Wuhan-Hu-1(NC_045512)基因组作为参考序列,使用CLC Genomics Workbench(Version 21.0)软件对测序原始下机数据进行序列拼接。
1.3.2 基于Ion Torrent S5平台的二代靶向测序建立基于Ion Torrent S5平台的新冠二代靶向测序方法,采用Thermofisher提供的新型冠状病毒全基因组捕获引物(242对引物)和SuperScript IV VILO Master Mix试剂盒(Thermofisher,美国)对提取的病毒RNA进行全基因组特异性扩增,扩增得到250-350bp大小不等的基因片段。扩增产物经回收后,按照Ion Torrent AmpliSeq Library Kit Plus(Thermofisher,美国)操作步骤构建测序文库,采用Ion OneTouch2系统构建测序模板。使用Thermofisher测序平台的 Ion Torrent S5测序仪和Ion530芯片进行全基因组深度测序。以SARSCoV-2 Wuhan-Hu-1(NC_045512)基因组作为参考序列,使用CLC Genomics Workbench(Version 21.0)软件对测序原始下机数据进行序列拼接。
1.3.3 基于MinION平台的三代靶向测序 建立基于MinION平台的新冠三代靶向测序方法,对提取的核酸使用针对SARS-CoV-2特异性引物(109对引物)进行靶向扩增,富集病毒基因组核苷酸序列片段,产物采用Agencourt AMPure XP磁珠进行纯化。使用Qubit3.0及Qubit dsDNA HS Assay Kit对纯化后DNA进行核酸浓度测定。利用连接法建库试剂盒对纯化后的核酸进行建库,并使用英国牛津Nanopore公司的MinION测序仪及R9.4.1 Flowcell测序芯片进行测序。使用Nanopore公司提供的artic-ncov2019分析软件对序列进行拼接。
1.4 全基因组序列分析 使用DNAstar软件对全基因组序列进行分析,与GISAID数据库中序列进行同源性比对,使用Mega软件对序列进行比对,并基于最大似然法构建系统进化树,参比序列来源于GISAID数据库和GenBank数据库。
2 结果
2.1 新冠肺炎病例的基本情况 本研究中的新冠肺炎病例分别为疫情初期间(2020年1月至2月)和外防输入期间(2020年3月至10月)的相关病例,其中疫情期间的病例为湖北输入及本地续发病例,共33例,来自江西省10个设区市,外防输入期间均为境外输入病例,共5例,分别来自美国、俄罗斯、菲律宾和刚果金等4个国家。38例病例年龄为18~79岁,男女分别为22人和16人。
2.2 全基因组测序 成功建立了SARS-CoV-2二代宏基因组测序、SARS-CoV-2二代和三代靶向测序方法,见表1。共获得所有病例共38条新冠肺炎确诊病例的临床样本全基因组,新冠全基因组序列长度为29867bp。所有序列均上传至GISAID和GWH数据库,序列号为EPI_ISL_421237-EPI_ISL_421254,EPI_ISL_421256-EPI_ISL_42126 2,EPI_ISL_455460-EPI_ISL_455467,WGS001712,WGS001721,WGS001722,WGS020746,WGS0189 64。

表1 三种方法比较
2.3 SARS-CoV-2病毒全基因组特征 通过构建的SARS-CoV-2病毒全基因组进化树发现,我省新冠病毒基因分属两个不同分支,分别为S型/A分支和L型/B分支,其中湖北输入及本地病例中有18例属于S型,15例属于L型,而境外输入病例均属于L型/B分支,经“Pangolin”分型分析,hCoV-19/China/Nanchang/JX216/2020为B.1分支,hCoV-19/China/Jiujiang/JX221/2020为B.1.1分支,hCoV-19/China/Nanchang/JX222/2020为 B.1.1.95分支,hCoV-19/Nanchang/JX554/2020为B.1.1.63分支,hCoV-19/China/Yingtan/JX572/2020为B.1.1.306分支。Blast比对显示,分别与hCoV-19/USA/NYNYUMC879/2020、hCoV -19/Switzerland/BS0914/2020、hCoV-19/USA/WA-UW138/2020、hCoV-19/Hong Kong/HKU-200723-102/2020、hCoV-19/Israel/CVL-n-6051/2020同源性最高。
以Wuhan-Hu-1(NC_045512.2)为参考基因组,对疫情期间的湖北输入及本地病例SARS-CoV-2核苷酸和氨基酸分析发现,33株SARS-CoV-2核酸变异数在1~6个之间,共检测到变异位点40个,除C8782T和T28144C连锁突变外,其余突变位点均只出现在1~3个病毒中,对应于6个编码区的39个氨基酸位点,其中ORF1ab编码区24个(61.54%),S编码区7个(17.95%),N编码区5个(12.82%),ORF3a、M、ORF8编 码 区 各 1个(2.56%);存在25个非同义突变(62.5%),其中S蛋白(刺突蛋白)存在6个非同义突变,见表2。未发现插入及缺失变异。
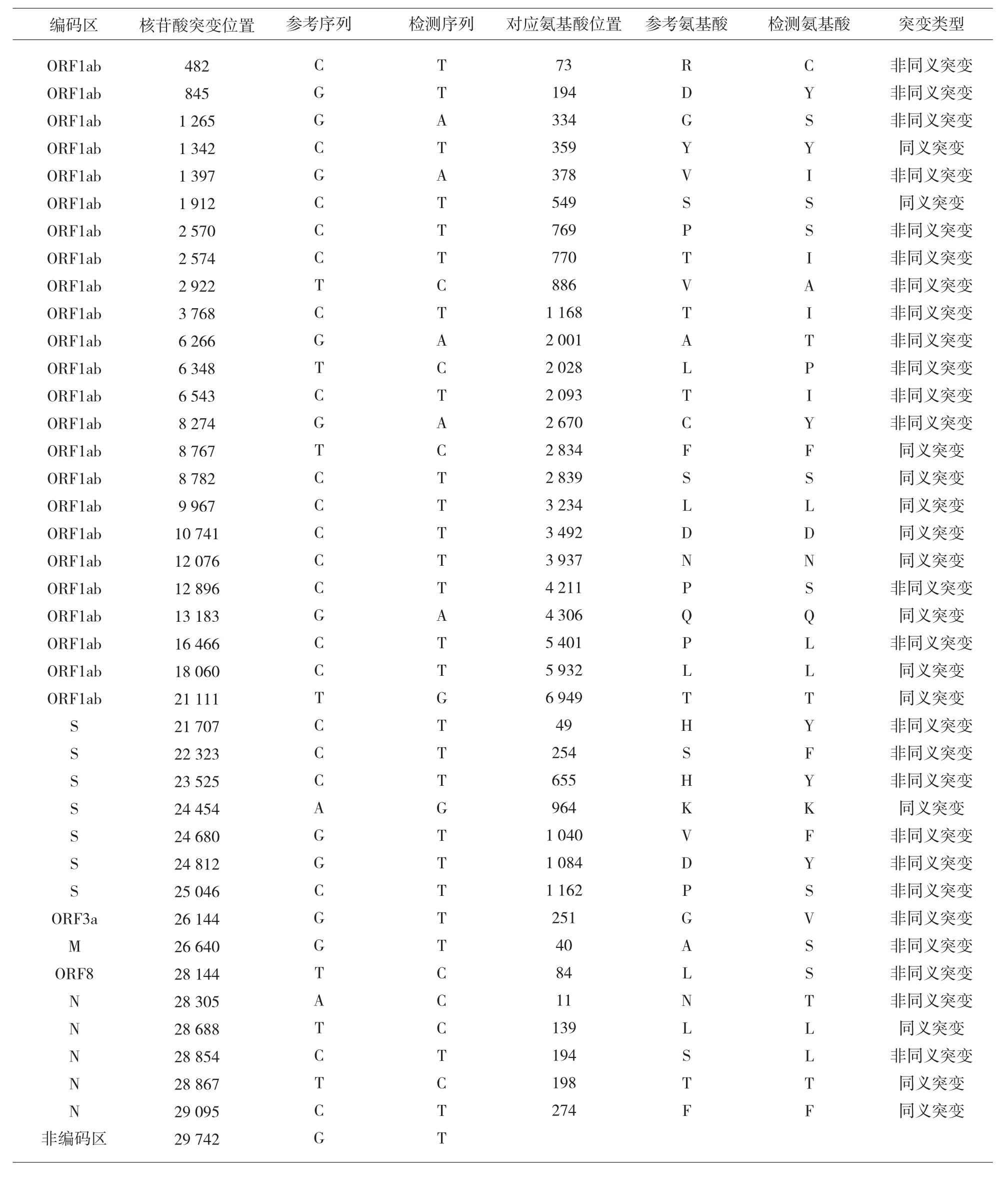
表2 湖北输入及本地病例新冠病毒氨基酸及核苷酸变异情况

图1 SARS-CoV-2全基因组核苷酸序列系统发育树
以Wuhan-Hu-1(NC_045512.2)为参考基因组,对外防输入时期的SARS-CoV-2核苷酸和氨基酸分析发现,5例病例基因组核苷酸变异数为7~15个,见表3。共检测到变异位点29个。其中ORF1ab编码区20个(51.28),S编码区2个(5.13%),N编码区5个(12.82%),非编码区2个(5.13%)。对应的氨基酸突变类型包括9个同义突变,18个非同义突变,见表4。和我省早期湖北输入及本地病例无相同变异位点。

表3 境外输入病例新冠病毒核苷酸变异位点

表4 境外输入病例新冠病毒氨基酸及核苷酸变异情况
3 讨论
新型冠状肺炎为新发传染病,SARS-CoV-2易发生变异,快速准确鉴定和分析SARS-CoV-2来源和基因变异特征,对于疫情防控具有重要意义。二代、三代测序技术在新型病毒的基因变异研究、病毒溯源等方面中具有巨大作用,无论是系统发育分析还是重要功能蛋白突变位点的识别都需要基于精准的全基因组序列信息[5]。本实验室在疫情初期通过建立了SARS-CoV-2二代宏基因组测序方法,对江西省第一例新冠肺炎病例的咽拭子样进行了全基因组测序,于2020年1月18日即获得了第一个新冠全基因组序列。由于宏基因组测序方法对样本中的所有核酸序列进行测序,因此获得的SARS-CoV-2基因数据不多,不仅测序时间长(36个小时),而且测序覆盖度较低,不适用于对SARS-CoV-2的精准测序。为了更精准和快速的对SARS-CoV-2进行基因组测序,本实验室在2020年2月和9月相继建立了基于Ion Torrent S5平台的新冠肺炎病例临床样本的二代SARS-CoV-2靶向全基因组测序方法和基于MinION平台的三代SARS-CoV-2靶向全基因组测序方法,获得全基因组测序最短时间为12 h,在外防输入期间江西省新冠肺炎病例的确诊和溯源工作中发挥非常重要的作用,为疫情防控争取了非常宝贵的时间,为江西省保持489 d(截止2021年6月30日)无新增本地确诊病例报告提供了实验室证据[13]。
对疫情期间临床样本的SARS-CoV-2的全基因组分析结果显示,发现江西省的新冠病毒全基因组序列与在Genbank数据库公布的第一条SARS-CoV-2参考基因组(NC_045512.2)的序列相比,变异数最多仅6个,且变异位置分散,提示疫情初期,江西省SARS-CoV-2基因相对稳定,变异率较低[6]。根据Tang等[7]早期提出根据C8782 T(ORF1ab:C8517T)和28144(ORF8:T251C)突变可将的SARS-CoV-2分为S型和L型两个型,2020年1月7日之前主要为S型,1月7日后,L型约占70%,S型占30%。根据该分型方法,本研究的33例病例有18株为S型,15株为L型,在疫情比较严重的设区市(南昌市、上饶市和新余市)均存在两种型别,推断我省疫情早期多为湖北输入及输入相关病例,与流行病学调查信息一致。
通过深入分析发现疫情期间刺突蛋白(S蛋白)上发生了6个非同义突变,S蛋白是介导病毒与宿主细胞膜发生融合并实现传播的表面糖蛋白,具有多个引起机体免疫反应线性抗原表位,是影响病毒传播及其致病性的重要蛋白[8-9]。根据国家基因组科学数据中心2019新型冠状病毒信息库对SARS-CoV-2基因组的变异注释,本研究中出现的6个位点变异的群体发生率均小于0.05,为较温和的突变[10],推测这些位点的变异对其传播性影响较小,并非可稳定遗传的变异,SARS-CoV-2在江西省内并未发生本地的广泛流行。
对境外输入病例全基因组分析结果显示,5例病例与我省早期病例变异位点并不相同,经blast比对,与国外同时期的病例同源性较高,与流行病调查结论一致,显示均为境外输入。根据” Pangolin” 分型法[11-12],5例病例的SARS-CoV-2分别属于B.1、B.1.1、B.1.1.63、B.1.1.95、B.1.1.306,不是世卫组织确定“关切变异株(VOC)”和“关注变异株(VOI)”。
综上所述,本实验室建立的三种临床样本的SARS-CoV-2全基因组测序技术,在新冠肺炎疫情不同阶段的防控工作中发挥了重要作用。在疫情初期阶段,证实了有效的公共卫生防控举措可以减少病毒的基因变异;在内防输入阶段,将检测周期缩短至12 h,在全基因组层面证实新冠肺炎确诊病例均为境外输入病例,及时为本土病例零增长、减少民众恐慌和维护社会和谐提供可靠的实验室证据。
