基于深度学习的金属离子-有机配体配位稳定常数的预测
2022-02-18齐书平王文龙张磊都健
齐书平,王文龙,张磊,都健
(大连理工大学化工学院,化工系统工程研究所,辽宁 大连 116024)
引 言
金属离子(Mn+)和有机配体(L)的络合过程是配位化学中应用最广泛的过程之一[1],涉及催化[2-4]、湿法冶金[5]、医疗[6]、生物医学与环境监测[7]等多种领域。该过程在这些领域的应用都离不开配合物的稳定性问题,而配合物的稳定常数是衡量溶液中有机配体与金属离子间结合稳定性的重要指标。目前,获取配合物稳定常数的传统方法是通过实验方法测定或通过分子模拟计算。实验方法包括电位法、分光光度法、色谱法等,这类方法通常烦琐复杂且耗时,特别是对大环化合物需要几天或几周的时间才能达到平衡。传统的分子模拟计算方法主要是利用密度泛函理论(density functional theory,DFT)或分子动力学(molecular dynamics, MD)进行几何优化和能量计算,通常费时费力且很难达到模型精度要求。
由于上述原因,利用实验数据建立回归模型预测金属离子和配体的稳定常数成为一种很有潜力的方法。此前,文献中报道过许多试图预测配体和金属离子间稳定常数的机器学习模型,涉及的机器学习方法包括多元线性回归(multiple linear regression, MLR)、支 持 向 量 机(support vector machine, SVM)、K-近邻(K-nearest neighbor, KNN)等[8-18]。这些模型中的绝大多数都是在某种确定的实验条件下,局限于构建单一金属离子与不同配体的定量结构性质关系(quantitative structure-property relationship, QSPR)模型[8-16]或某一类有机配体及其衍生物(如席夫碱及其衍生物)与不同金属的QSPR模型[17],配体使用的大都是子结构片段(substructural molecular fragments,SMF)描述符,每个模型所用的数据集很小(几十到上百),配体的类型十分有限,也没有考虑金属离子和配体间的相互作用。针对这些模型缺陷,Chaube 等[18]提出了考虑不同实验条件的预测镧系金属离子-配体间稳定常数的机器学习模型,使用的描述符为物理化学描述符和拓扑描述符,该工作的不足之处在于模型只适用于性质相近的镧系金属离子,且没有考虑多配位的情况。综上所述,目前提出的模型仅针对有限类别的配体或金属离子而构建,且由于这些限制,这些模型的错误率较高,泛化能力较弱,限制了它们在差异很大的配体化学空间中的应用。
此外,传统的QSPR 模型通常基于成百上千个已知描述符的组合来提取分子特征[18],缺少对配合物的针对性。至于配合物稳定常数lgK的大小,主要与有机配体和金属离子形成的配合物的结构有关。而不同的金属离子和配体由于配位数和配位原子数的不同,难以表示整个配合物的分子图。因此为了便于建模,把每种配合物的表示分成了两步,第一步通过有机配体的分子图及原子和键的性质,准确地描述有机配体(包括顺反异构、手性等),模型认识了这个有机分子的特征,就有了预测的基础;第二步将多头图注意力网络(multi-head GAT)[19]提取的分子特征拼接金属离子的独热编码,考虑有机配体和金属离子的相互作用。
由于稳定常数还受温度、介质、离子强度等实验条件的影响[20],因此本研究除了考虑有机配体和金属离子的相互作用,还编码了稳定常数测定时所处的实验条件。本文提出的深度学习模型以配体的简化线性输入规范(simplified molecular input line entry system, SMILES)转换得到的分子属性图作为输入,经图注意力特征提取层,识别对配合物稳定常数有重要贡献的节点特征,得到分子图的特征表示并将提取的分子特征与使用独热编码的金属离子和实验条件进行拼接后送入全连接层,预测金属离子与有机配体的稳定常数。
1 基于深度学习的配合物稳定常数预测模型框架
本文提出的模型框架由数据准备和模型训练两部分组成,如图1 所示。首先,从IUPAC(the international union of pure and applied chemistry,IUPAC)稳定常数数据库[21]的子库(mini stability constant database)中获取建立模型所需的实验数据,利用 DGL[22](deep graph library,DGL)对有机配体生成分子属性图,其中各原子的属性值由RDKit[23]计算得到。其次,使用独热编码对实验数据中的金属离子和实验条件部分进行编码,获得条件特征向量。最后,将分子属性图送入建立的多头图注意力网络进行分子特征的抽提,将所得特征与条件特征向量进行拼接,送入全连接层进行稳定常数的预测。

图1 预测金属离子-有机配体稳定常数的深度学习模型框架Fig.1 A deep learning model framework for predicting the stability constants of metal ions-organic ligands
1.1 数据准备
本文使用IUPAC SCDatabase 的子库mini-SCDatabase作为训练所需的实验数据来源。数据的提取过程为:首先,根据所研究的金属离子的种类选定金属离子下载一个文本文件,文件中每条数据包括配体名称(俗名和配体的大致分类,部分数据包含CAS 号,分子式)、金属离子、介质的种类和浓度、实验温度、稳定常数;然后,编写python脚本自动整理数据,删去不含碳和碳数大于20 的配体,从文本文件中提取建模所需的信息并导出到excel 表格中,包括配体名称、CAS 号、实验条件、单配位 (Mn+∶L = 1∶1)稳定常数lgK1和双配位(Mn+∶L=1∶2)稳定常数lgβ2;最后,根据配体的CAS 号在PubChem 数据库中爬取对应的SMILES,对CAS 号错误或缺失的配体,根据分子式和俗名手动写出SMILES。对于本文所涉及的29种金属离子,经筛选和整理后得到7127条实验数据,每条数据由有机配体、金属离子、实验条件(实验介质、支持电解质的离子强度或混合溶剂的体积比、配位状态以及实验温度)和稳定常数(配位比为Mn+∶L = 1∶1 的lgK1和配位比为Mn+∶L=1∶2的lgβ2)组成。数据集中络合物的金属离子和稳定常数的数据点分布如图2所示。从图2(a)中可以看出除少数金属离子外,大多数金属离子的数据量都大于100,从图2(b)中可以看出稳定常数大都处在(1,15)的取值范围中。
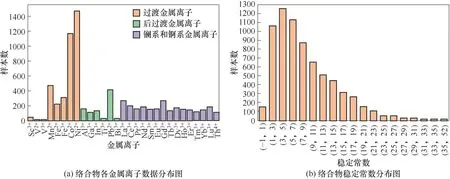
图2 数据分布图Fig.2 The distribution of data points
本文使用DGL 对所有实验数据中涉及的1371个有机分子生成分子属性图,属性图中原子及键的特征与编码方式如表1所示。分子属性图不仅考虑了原子层面的特征(如原子种类、形式电荷、原子杂化方式等)和分子层面的特征(如Gasteiger 电荷、Crippen 摩尔折射率、拓扑极性表面积TPSA 等),还考虑了各种化学键的特征(如键类型、键方向、键数量等)。原子特征的选取参考了Coley 等[24]在分子属性图方面的工作,并在原子特征矩阵中引入键特征参数,既考虑了分子的空间信息,也能更有效地表示原子所处的化学环境。

表1 分子属性图中原子与键的特征及编码方式Table 1 Features and coding methods of atoms and bonds in molecular attribute diagrams
1.2 模型训练
本文在模型训练过程中采用的是留出验证,即把数据划分为训练集、验证集和测试集,划分比例为90∶5∶5,其中验证集用来选择模型的超参数(如学习率、层数等),测试集用来评估模型性能。此外,本文在构建训练集时充分考虑了有机配体的多样性,具体做法为:首先,在随机打乱的数据池中,顺序依次取出所有第一次出现的有机配体SMILES所对应的数据,共1371 条数据,然后在剩余的数据中随机抽取5044条数据共同组成训练集。最后,随机抽取验证集数据356条,剩下的则放入测试集。
深度学习模型主要由多头自注意力特征提取层和全连接层两部分组成。其中GAT 层作为第一层次读入分子属性图,将其按照图神经网络节点信息更新规则对分子属性图进行迭代,使得模型认识到这个有机配体;全连接层作为第二层次读入经过抽提的分子属性图、金属离子以及实验条件,目的是让在模型认识到这个有机配体的情况下再进一步获取其他必要信息,以便进行lgK的预测。之所以将分子属性图单独作为GAT 层的输入而不结合其他信息(如金属离子或某些实验条件),是因为希望所建立的模型先通过属性图了解到分子的信息后大致确定一个预测范围(也就是配体络合能力的强弱),再通过后续输入的其他必要信息缩小预测范围,直至实现精准预测。综上,通过建立双层次的图神经网络,模型具有了学习预测lgK所需要的物理知识的能力。多头自注意力特征提取层是通过multi-head GAT 实现的,它背后的关键思想是注意力机制,可以将不同的权重分配给邻域中的不同节点,特征提取过程后得到的信息对稳定常数预测更为有效。计算如式(1)所示。

本文中多头图注意力机制的实现使用了DGL框架(后端为PyTorch[25])。DGL 是一款易于使用的开源图神经网络计算框架,它通过与主流的深度学习框架(目前支持Pytorch、TensorFlow[26]等)集成,实现了从传统的张量运算到图运算的自由转换。本文使用DGL 库内置的GATConv 模块搭建8 层的多头图注意力网络对分子属性图进行特征提取,将提取的分子特征与使用独热编码的金属离子与实验条件进行拼接,送入3 层的全连接层进行配合物稳定常数的预测。全连接层的大小分别为128、32 和1。训练过程中的批次大小设置为128,使用初始学习率为0.0003 的Adam 优化器,在训练1500 代后停止。
2 训练结果与案例分析
2.1 模型训练结果
由于本文的稳定常数预测是一项回归任务,故选用均方误差(mean square error, MSE)作为损失函数,如式(2)所示。
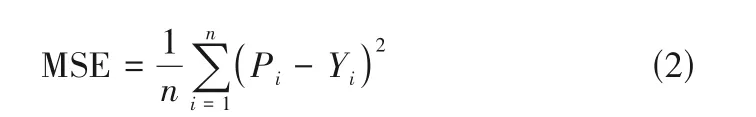
式中,Pi和Yi分别是第i个数据的预测值和实验值;n为样本总数。模型的训练结果如图3 所示,图3(a)为损失函数下降曲线,其中红色曲线表示训练集的损失函数曲线,蓝色曲线表示验证集的损失函数曲线。从图3(a)中可以看出在第1500 代时训练集与验证集损失函数趋于稳定,故停止训练。模型的预测能力可用回归决定系数R2和均方根误差(root mean square error, RMSE)进行评价。R2计算如式(3)所示。RMSE计算如式(4)所示。

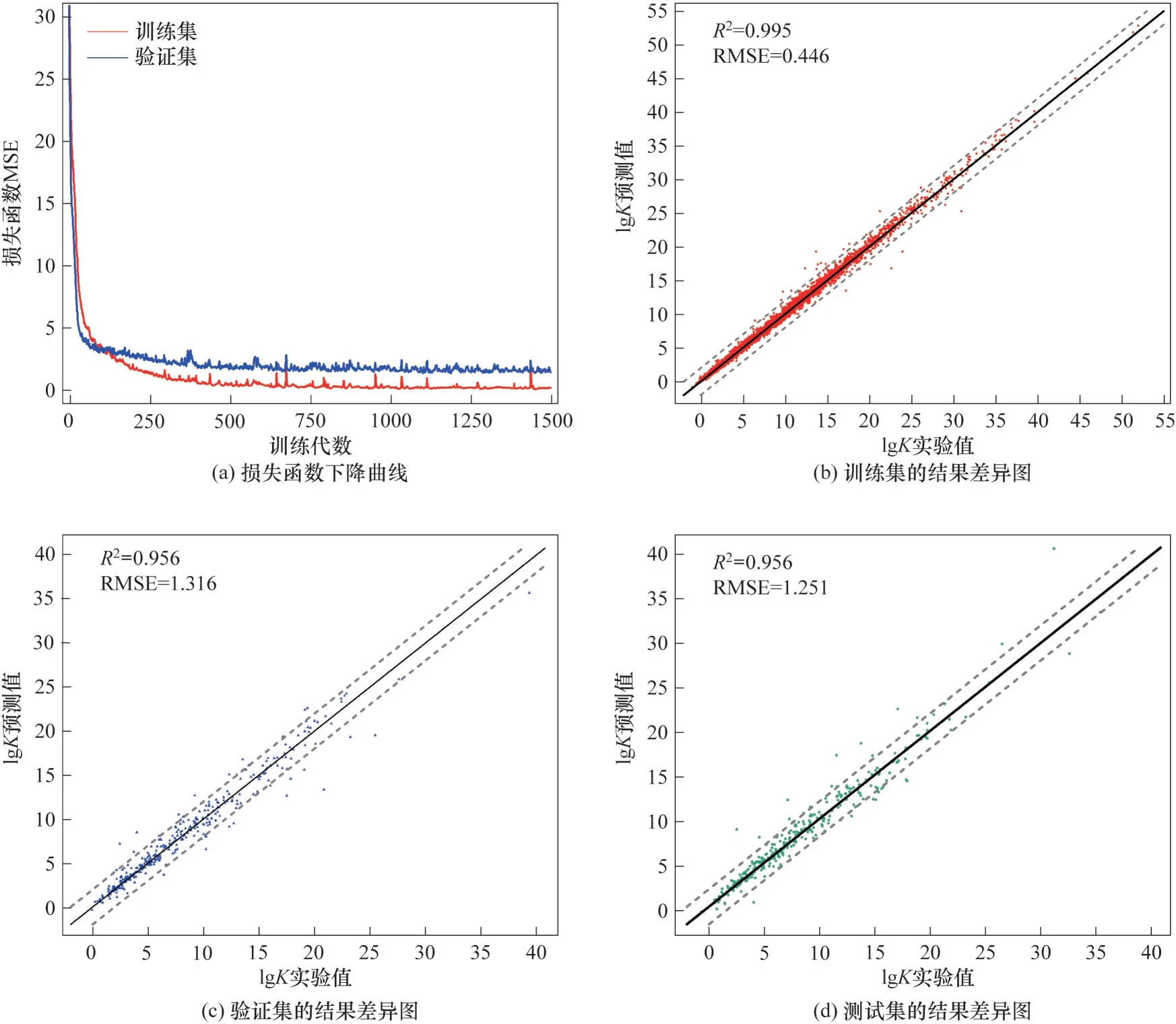
图3 模型训练结果Fig.3 Model training results
图3(b)、(c)、(d)分别为训练集、验证集和测试集的实验值和预测值之间的结果差异图,其中位于两条虚线之内的点表示其与实验值的绝对误差不超过2。训练集、验证集和测试集的R2与RMSE 分别为0.995/0.446、0.956/1.316 和0.956/1.251。数据集中共有88 个点在误差线外,其中训练集中有29个在误差线外的数据点,占训练集总样本数的0.45%;验证集中有30 个点在误差线外,涉及13 种金属离子,占验证集样本总数的8.42%;测试集中有29 个点在误差线外,涉及14 种金属离子,占测试集样本总数的8.14%。在这88 个点中,稳定常数的实验值大于15 的点有41 个,占这个区间所有数据点的3.90%,实验值小于15 的点占同区间所有数据点的0.77%,这是由稳定常数的数据分布导致的。
2.2 案例分析
近年来,研究者们广泛使用DFT 计算[27-28]、MD模拟[29-30]来获得配合物在溶液中的络合自由能ΔG,络合自由能和稳定常数间的转化如式(5)所示[27]。
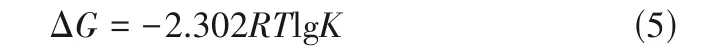
式中,R为热力学常数,R= 8.314 J·mol-1·K-1;T为热力学温度。这些方法虽然对比黑箱的深度学习模型解释性更强,但想要得到可靠的结果,需要选择合适的基组或力场,并设置超参数等,对不同的体系计算方法也不同,因此找到合适的计算方法是一个难题。以DFT 计算为例,定量计算配合物在溶液中的络合自由能需要使用计算步骤烦琐的热力学循环。此外,即使使用精度较高的基组(如PBE/TPZ),计算结果往往也不能尽如人意,大多情况下只能定性比较[27]。
因此,本节使用前文建立的深度学习模型来预测文献中螯合物的稳定常数,表2 为模型预测结果和文献中DFT 计算结果的对比,结果展示了模型强大的预测能力,所选用的配体和金属离子对在数据集中没有出现过。预测结果表明,本文建立的深度学习模型计算成本更低,泛化程度更高,更容易建立与应用,且在大多数情况下模型预测结果更为准确。表2所示的预测情况中,第三种配体与Ni2+的单配位稳定常数预测误差最大,绝对误差为4.71,这是因为SCDatabase 数据库不再提供维护,所以本文所用为其子数据库mini-SCDatabase,数据量不够丰富,稳定常数较大的数据点较少,存在数据不平衡的问题。以单配位态的Ni2+为例,数据集中仅有4%的数据lgK1>17,模型学习到螯合能力强的配体的特征较少,以致于模型对螯合能力太强的配体预测偏差较大。
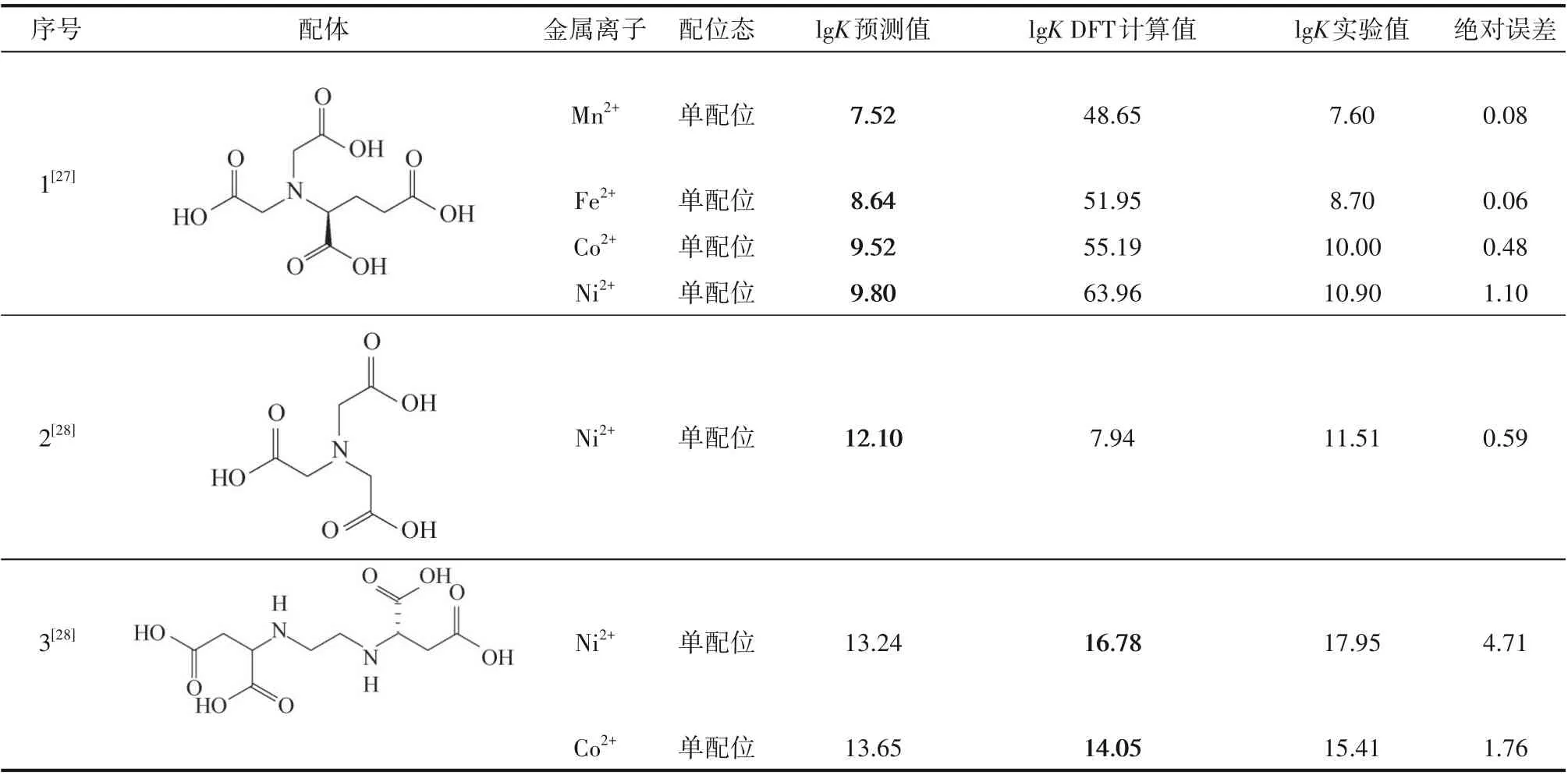
表2 模型预测结果和DFT计算结果与实验值的比较Table 2 Comparison of model prediction results and DFT calculation results with experimental values
3 结 论
本文利用基于多头图注意力网络对29 种金属离子和1371 个有机配体构建配合物稳定常数预测模型,该模型综合考虑了多种影响因素,可对多种配体、多种金属离子、多种实验条件进行稳定常数的高通量预测。本文的主要贡献如下:(1)创新性地提出了一种配合物的表征新范式,并将multihead GAT 用于配合物的性质预测;(2)本文模型涉及的金属离子和有机配体的多样性远超以往研究,通过建立一个模型完成了对过渡金属离子(Sc3+,V2+, V3+, Mn2+, Fe2+, Fe3+, Co2+, Ni2+)、后过渡金属离子(Al3+,Ga3+, In3+, Tl3+, Pb2+, Tb3+, Bi3+)、镧系和锕系金属离子(La3+, Ce3+, Pr3+, Nd3+, Sm3+, Eu3+, Gd3+, Tb3+, Dy3+,Ho3+, Er3+, Tm3+, Yb3+, Th4+)与有机配体的稳定常数预测,模型适用范围广泛。
模型首先基于实验数据涉及的有机配体生成分子属性图,然后使用独热编码对金属离子和实验条件进行编码,最后将分子属性图送入特征提取层进行特征提取,将提取出的特征拼接上金属离子和实验条件的特征编码,送入全连接层预测稳定常数。模型在测试集上的RMSE 为1.251。与DFT 计算结果相比,本文提出的模型更为高效。而且值得注意的是,与仅针对螯合剂建模的模型不同,本文中参与建模的有机配体不仅有螯合剂(多齿配体),还有非螯合剂(单齿配体),这在很大程度上拓展了模型的可用范围,可用于对特异金属螯合剂的高通量筛选。此外,在本模型中使用多头图自注意力网络直接处理有机配体的分子图,可自动提取与所预测性质相关的分子特征,因此相信该方法还可以应用于稳定常数以外的其他配合物性质预测(如磁性、颜色等)。
