CeO2/CuFe2O4氧载体CH4化学链重整耦合CO2热催化还原研究
2022-02-18王保文张港刘同庆李炜光王梦家林德顺马晶晶
王保文,张港,刘同庆,李炜光,王梦家,林德顺,马晶晶
(1 华北水利水电大学能源与动力工程学院,河南 郑州450045; 2宁夏大学省部共建煤炭高效利用与绿色化工国家重点实验室,宁夏 银川 750021)
引 言
天然气作为主要的化石能源之一,储量巨大、清洁高效、应用广泛,近年来备受关注。根据国际能源署2022年预测,2040年全球天然气需求量将增长30%。随着能源利用与环境保护之间矛盾日益尖锐,天然气在未来将会有更大的应用市场[1]。天然气主要成分为CH4,与CO2并列为两大主要温室气体,当前大气中CO2浓度不断增加,全球气候变暖日益严重。尽管碳捕获和封存技术有助于减少大气中CO2排放量,但对CO2的综合利用能力有限,迫切需要开发CH4、CO2两大温室气体协同利用、高效转化的新技术和工艺[2]。
相对于利用效率较低的CH4直接燃烧技术,CH4重整制取合成气的间接转化技术更受关注[3]。所制成的合成气可通过费托合成制取液体燃料、甲醇等化工产品,更具前景且用途广泛。CH4水蒸气重整是目前唯一商业化的技术,但该技术为强吸热反应、能耗高,合成气氢碳比不适合下游化工过程应用[4];而CH4干重整同样为强吸热反应,反应所需温度高,易造成催化剂积炭失活[5];尽管CH4部分氧化反应可以制备H2/CO 比值接近2 的合成气,直接应用于下游费托合成过程,但该反应速率快,需要纯氧参与,既增加制氧成本又存在爆炸风险[6]。
由于CO2等大量排放导致的温室效应日益严重,化学链与CO2资源化利用相结合的工艺技术开始受到密切关注。Najera 等[7]在化学链燃烧技术的基础上进一步创新性提出了CH4化学链干重整技术(chemical looping dry reforming,CLDR),类似于化学链燃烧(CLC),但CH4不是完全燃烧产生CO2、H2O,而是被氧载体(OC)部分氧化生成合成气(H2+CO),主要包括CH4部分氧化、CO2催化还原及空气氧化再生三个反应步骤:


图1 甲烷化学链干重整制合成气工艺Fig.1 Chemical looping dry reforming (CLDR) of methane technology for syngas
当前CLDR 的主要挑战是发展对H2和CO 具有高选择性、易于被CO2再生、储氧能力和氧迁移率高且热稳定性强的氧载体。CO2分子结构极为稳定,由于其反应活性小且转化率低,因此在活化或分解时面临着巨大的挑战。CO2裂解不仅需要能量输入,还需要合适的催化剂[8]。当前,在Fe、Mn、Cu、Ni、Co、Ce 等[9-13]单金属氧载体中,Fe 基氧载体因载氧能力高、价格低廉、无潜在二次污染等优势得以广泛应用,指出铁氧化物只有深度还原到不高于Fe3O4的低价态,才能对催化CH4重整及CO2转化起作用[14],但Fe与FeO塔曼温度低,在高温下连续反应时会烧结失活;同时,鉴于Fe2O3与CH4的低反应性而Fe 对CH4分解积炭的高活性,需要进一步活化改性提高其反应活性与稳定性[15]。相对于Fe 基氧载体,Cu 因其低温反应活性高、价格低廉且对CH4裂解积炭不起作用被认为是良好的改性金属。但是,铜基氧载体由于其极低的塔曼温度,容易烧结,一般不单独使用[16]。
与单一金属氧化物相比,复合金属氧化物可以通过调控化学计量比,产生更多缺陷来增加氧扩散,具有更好的反应活性和稳定性[17]。Qin 等[18]研究表明Cu 的掺杂促进了Fe2O3氧载体中氧空位的形成能力,显著降低了CH4活化能垒,可以有效提高Fe基氧载体的反应性能。因此,Fe2O3向Fe3O4转化倾向于燃料燃烧,而Fe3O4的深度还原则有利于CO 产生。Yüzbasi 等[19]通过H2原位脉冲实验证明Cu 的添加使FeO 还原为金属Fe0的速度加快了6 倍。反之,Fe 助剂的加入也可以显著提高Cu 催化剂的活性及稳定性[20]。而Wang 等[21-22]将CuFe2O4应用于煤化学链燃烧,证实是一种很有前途的氧载体,具有比单一CuO或Fe2O3更高的反应活性、良好的抗烧结性能以及广泛的燃料适应性。Kang 等[23]将CuFe2O4应用于化学链重整制氢研究发现,Cu的生成可有效抑制CH4的裂解并缓解积炭,增强了还原反应动力学,降低了还原温度,与Fe3O4相比,CuFe2O4有着较短的反应时间、低炭沉积和高CO选择性。
另外,CeO2因其良好的储放氧特性和离子传递能力、优异的抗积炭性能、循环稳定性且与CH4反应具有较高的CO 选择性[24],引起了研究者的广泛关注。Wei 等[25]把CeO2改性的Fe2O3用于生物质热解气化学链重整耦合CO2裂解反应中,发现CeFeO3固溶体的形成有助于促进氧迁移并产生氧空位[26],而CeO2的添加增强了金属间的相互作用和反应性能,氧迁移率也通过连续的氧化还原反应得以改善,从而抵消了烧结带来的不利影响。同时,改性的CeO2还具有将CO2催化转化为CO 的潜力,可用于CO2资源的有效利用[25,27],但CeO2较高的还原温度和较低的合成气产量成了制约CeO2氧载体工业应用的最大障碍。为了提高CuFe2O4的反应性和CO 选择性,Kang 等[28]在ZrO2和CeO2上负载CuFe2O4,然而这种方法导致CuFe2O4的含量减少到仅有20%(质量),实际应用方面经济性较低。
本文采用溶胶-凝胶燃烧合成法一步制备不同质量掺杂比的CeO2/CuFe2O4复合氧载体,充分利用其中CuFe2O4氧容量高、对CH4反应活性突出、抗烧结能力强以及CeO2的良好氧传递特性和循环稳定性,对其部分氧化CH4耦合热催化还原CO2的复合性能加以系统研究和定量评估,重点关注不同CeO2掺杂量对氧载体反应性能的影响,并通过连续氧化还原循环实验以证实该复合氧载体的CH4转化率及CO2催化还原能力。
1 实验部分
1.1 氧载体制备
所需材料,试剂级硝酸铜[Cu(NO3)2·3H2O,AR,> 99%]、硝酸铁[Fe(NO3)3·9H2O,AR,> 99%]、硝酸铈[Ce(NO3)3·6H2O,AR,> 99%]、尿素(CON2H4)、聚乙烯醇(PVA)均购置于国药化学试剂有限公司。采用溶胶-凝胶燃烧合成法一步制备不同掺杂量的CeO2/CuFe2O4氧载体。首先称取一定质量的硝酸铜、硝酸铁、硝酸铈、尿素加入预先配制好的PVA 和去离子水中,分散均匀后置于75℃恒温水浴中,并利用电动搅拌器匀速搅拌直至形成果冻状湿凝胶,再放进鼓风干燥箱中80℃及120℃下分步干燥形成干凝胶;之后在600℃的电炉中点火直至所有干凝胶自蔓延燃烧并形成疏松多孔的点火产物,最后在950℃的马弗炉中以空气气氛恒温煅烧2 h以提高氧载体的机械强度。研磨、筛分出粒径在105~300 μm的氧载体备用。制备的氧载体中,CeO2掺杂量从0、10%、20%、30% 到40%,分 别 命 名 为100CF、10Ce90CF、20Ce80CF、30Ce70CF、40Ce60CF,而单独的 CeO2命名为100Ce。
1.2 氧载体表征
氧载体的晶相组成在日本理学公司生产的Rigaku SmartLab 9 kW 型X 射线衍射仪(XRD)上进行测定。仪器参数为40 kV、40 mA 的Cu Kα射线,λ=0.1542 nm,扫描速度8(°)/min,扫描范围2θ=10°~90°。
氧载体的氧传递特性采用H2-TPR 方法在北京彼奥德PC-1200 化学吸附仪上进行测试。将100 mg 的氧载体置于U 形石英反应管中,先在Ar 气氛中于300℃恒温2 h 进行反应前预处理,冷却至室温后,切换至10%(vol)H2/Ar 混合气,流速为50 ml/min,采用10℃/min的升温速率从室温升至1000℃。
氧载体的结构特性采用北京精微高博JWBK100比表面积及孔径分析仪进行测定。样品先在180℃下脱气预处理2 h 以上,再在液氮(-196℃)下进行吸、脱附测试。
氧载体的形貌特征采用Thermo Fisher Scientific公司扫描电镜Apreo 2C 分析。取少量样品沾到导电胶上,放置于样品台上观察样品不同倍率下的微观形貌,而组分分布则采用仪器联用的EDX 加以测试。
最后,氧载体界面元素分布在Thermo Fisher Scientific 公司 Nexsa 型X 射线光电子能谱分析仪(XPS)测定,单色Al 靶(E=1486.68 eV)发射源,工作电压12 kV,工作电流6 mA,通道能量为50 和200 eV,数据处理采用仪器自带Avantage软件进行。
1.3 固定床实验
多功能立式高温固定床装置如图2 所示,其中反应器为长800 mm、内径30 mm 的石英管,由电炉加热,最高反应温度可达1200℃,进出口采用橡胶密封圈密封,推拉杆由多孔布风板侧伸入反应器内部,通过可上下移动的推拉杆来确定样品所在反应器中的位置以灵活调整反应温度。实验时,先在布风板上放置一定量石英砂,再称量并放置定量氧载体。实验时,先在N2气氛中程序升温至设定温度后,关闭N2,再通入1 L/min的10%(vol)CH4/N2混合气实现CH4部分氧化;然后在CO2还原阶段,通入1 L/min的10%(vol)CO2/N2混合气以实现CO2的还原并部分再生被还原氧载体;最后,再通入1 L/min 空气在等温条件下实现部分氧化的氧载体充分氧化再生。实验过程中,采用气体分析仪(GASBOARD-3100)对尾气进行在线连续分析。
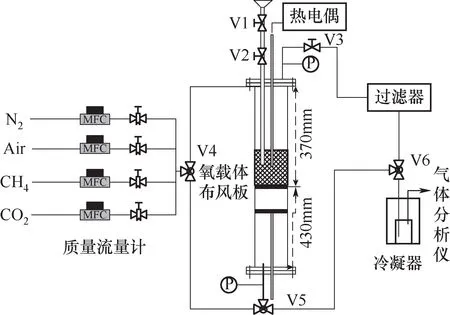
图2 固定床示意图Fig.2 Schematic diagram of fixed-bed reactor
1.4 氧载体性能评价
为了全面研究CeO2/CuFe2O4氧载体部分氧化CH4、还原氧载体在CO2气氛中的部分氧化及与空气的充分氧化再生,主要设计并发展了如下评价指标。
对于黏性情形, 计算域及网格点不变, 取Re=400, 800, 1 600, 3 000, 这是一组低分辨率数值模拟. 将原始WENO-CU6-M2格式的改进B的结果与已有LES模型对比, 如动态Smagorinsky模型和Hickel等[21]的自适应局部反卷积模型(adaptive local deconvolution model, ALDM), 且Brachet等[22]的直接数值模拟(direct numerical simulation, DNS)数据用作参照解.

式中,nCOFR,out、nCO2,FR,out、nH2,out分别为燃料反应器出口的CO、CO2、H2物质的量。
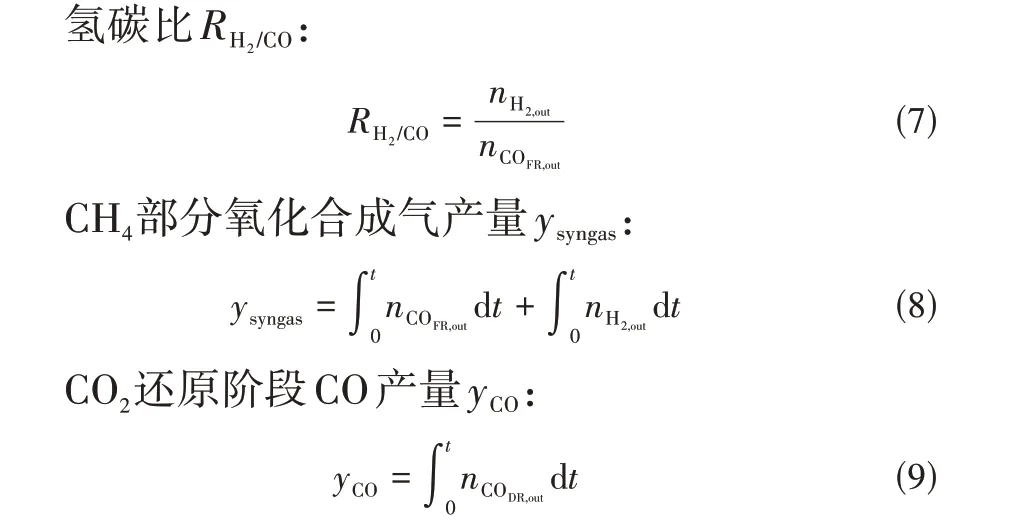
式中,nCODR,out为CO2反应器出口CO的物质的量。
2 实验结果与讨论
2.1 氧载体表征
图3 显示了新鲜氧载体的XRD 图,所有掺杂改性氧载体均可以检测到四方晶系尖晶石结构的CuFe2O4(JCPDS 34-0425)衍射峰,其中最强的衍射峰28.549°、35.861°分别对应萤石结构CeO2(JCPDS 43-1002)的(111)晶面和CuFe2O4(JCPDS 34-0425)的(211)晶面。随着CeO2掺杂量从0 增加到40%,CuFe2O4特征峰逐渐减弱,而CeO2特征峰逐渐增强,这与Selvan等[29]的结论一致,尽管他们的研究中CeO2在CuFe2O4中的掺杂量不大于20%。同时,尽管部分掺杂的CeO2可能会溶入CuFe2O4尖晶石中,但对CuFe2O4晶体结构总体上没有显著的影响且一直保持四方结构。因此,不同含量的CeO2掺杂入CuFe2O4中,其物相通过XRD 识别,仍为独立的CuFe2O4以及CeO2物相,可推测已经形成复合材料[30]。
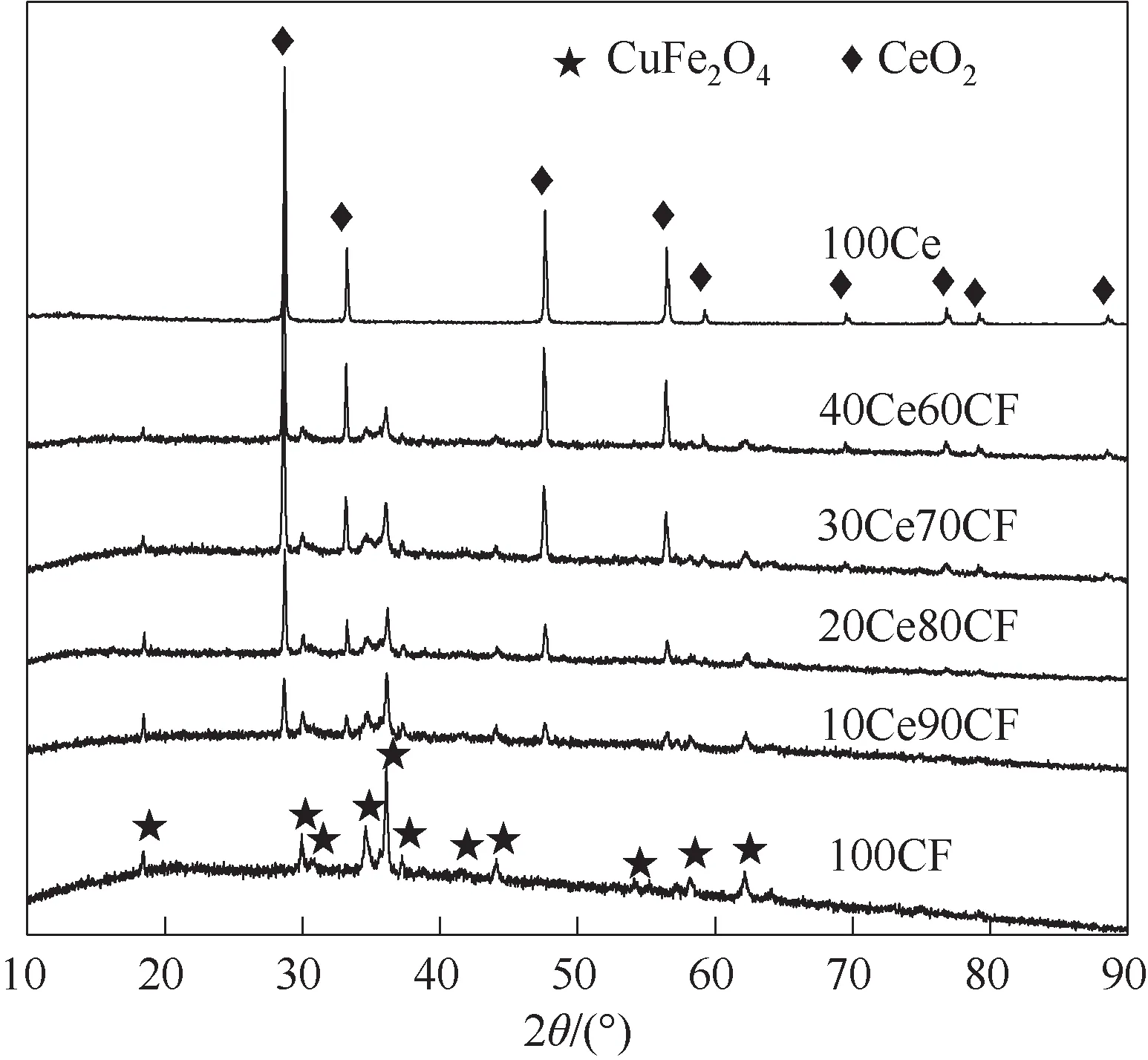
图3 自制氧载体的XRD谱图Fig.3 XRD patterns of the freshly prepared oxygen carriers
表1 分别为不同CeO2含量的复合CuFe2O4氧载体比表面积、孔径、孔体积等结构参数及其中主要独立相CuFe2O4和CeO2的晶粒尺寸。由表1 可知,CeO2掺杂后复合氧载体比表面积均大于独立的100CF 与100Ce 参考氧化物,且随掺杂量增加比表面积与孔体积均增大,40Ce60CF比表面积(5.820 m2/g)远大于独立的100CF(1.166 m2/g)与100Ce (2.262 m2/g)。较大的比表面能提供更多的反应活性位点,使得氧载体展现出更高的氧化还原性能。而CeO2掺杂氧载体孔径相较于100CF 变化不大,但远小于100Ce;同时,随着CeO2的掺杂,复合氧载体孔体积逐渐增加,较大的孔体积有助于提高氧载体气固反应中气体反应物及产物的传递。最后,复合氧载体中主要的独立相CuFe2O4、CeO2晶粒尺寸基于谢乐公式进行估算,如表1 所示,随着CeO2掺杂量的增加,鉴于掺杂的CeO2的位阻效应,CuFe2O4晶粒团聚程度得以有效抑制,其晶粒尺寸逐渐降低,在掺杂的CeO2中具有较好的分散性;相应地,所掺杂的CeO2晶粒也比单独制备的参考CeO2晶粒尺寸稍小,但当CeO2掺杂量大到40%时,由于其中CeO2晶粒的聚合,掺杂CeO2晶粒尺寸显著增大到49.80 nm,接近独立的参考CeO2的晶粒尺寸。
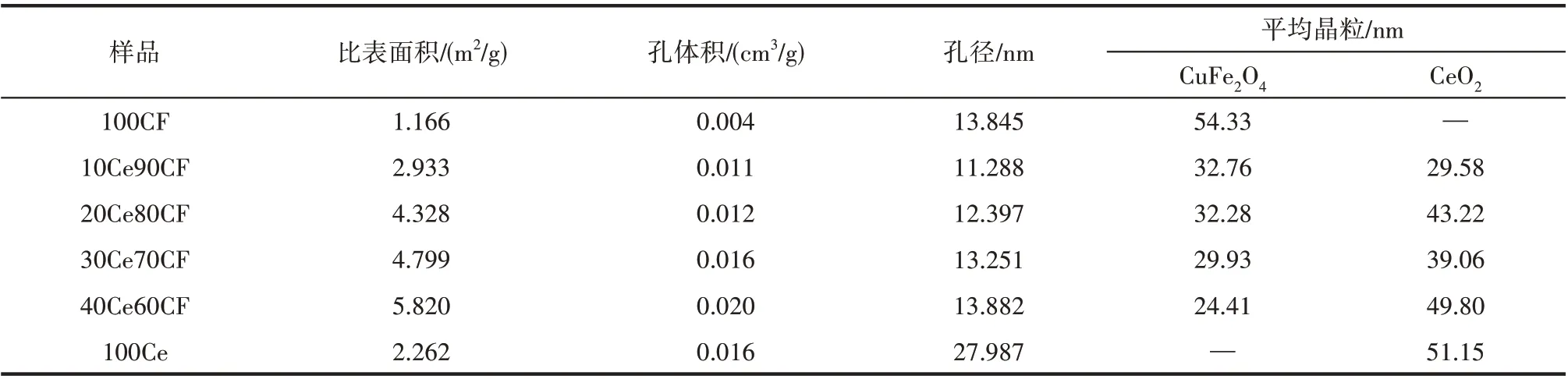
表1 氧载体物理结构参数Table 1 Physical structure parameters of oxygen carrier
图4(a)为所制备氧载体的N2吸附-脱附等温线,根据国际纯粹与应用化学联合会对N2吸附等温回线的分类[31],所有氧载体均显示具有H3型回滞环的Ⅳ型N2吸附-脱附等温线,表明制备材料主要由介孔结构组成,而H3 型回滞环等温线没有出现明显的饱和吸附平台,表明孔结构很不规整,为片状粒子堆积形成的狭缝孔,同时有大孔和非孔结构存在。进一步,从孔径分布图4(b)可以看出所制备氧载体孔径主要集中在2~10 nm。

图4 氧载体N2吸附-脱附曲线(a)和孔径分布(b)Fig.4 N2 adsorption-desorption curves of oxygen carriers (a) and pore size distribution (b)

图5 自制氧载体H2-TPR谱图Fig.5 H2-TPR profiles of the prepared oxygen carriers
图6为氧载体SEM图像,均采用溶胶-凝胶燃烧合成法一步制备,以30Ce70CF 为例,采用能谱分析仪测定Ce 组分分布,参见图6(h),可以看出CeO2较为均匀地分布在CuFe2O4中,随着CeO2掺杂含量的增大,氧载体孔隙结构更加发达,孔径与纯CuFe2O4相比略有增加,孔分布更加均匀,且氧载体表面团聚减少,呈现薄片状,具有较大表面积,这与表2 中BET 结果相一致。进一步,以30Ce70CF 为例,由图6(h)~(k),从EDX可以看出所制备的氧载体各元素分布均匀,完全达到预期效果。

图6 氧载体表面SEM-EDX图像Fig.6 SEM-EDX images of oxygen carrier surface
2.2 固定床CH4部分氧化恒温反应及其性能定量评估


图7 CuFe2O4-CeO2氧载体900℃下与CH4部分氧化气体产物分布Fig.7 Gas products distribution of methane partial oxidation with CuFe2O4-CeO2 at 900℃
图8 为氧载体在CH4部分氧化进行到5、10、20、30 min 时各气体产量分布。由图8(a)可知,在5 min时,只有40Ce60CF 与CH4部分氧化时合成气氢碳比达到2,但此时合成气产量较低,而持续反应到10 min 时20Ce80CF 与30Ce70CF 既具有较高合成气产量又可以保持合成气氢碳比约为2。此后,随着反应继续,CH4裂解加剧,氢气过量,合成气产量增加,但CH4裂解产生的积炭会覆盖氧载体活性位点,造成氧载体反应活性降低甚至失活。掺杂改性后的氧载体在CH4部分氧化反应初期释氧量远高于100CF,初期H2和CO 产量增加的同时CO2生成量降低,说明CeO2的掺杂使得氧载体有较多可用晶格氧,并且可有效提高氧载体的氧迁移能力,这与上述H2-TPR 分析结果一致。随着反应时间的延长,鉴于CeO2与CuFe2O4之间的协同作用,如图8 所示,合成气产量随CeO2掺杂量增加而增加;而 CeO2过量掺杂后,如表1 所示,CeO2晶粒团聚程度增加、晶体粒径加大,CeO2晶粒不能与CuFe2O4充分接触,其氧传递能力降低。因此,30Ce70CF 与CH4部分氧化时具有最高的合成气产气量。
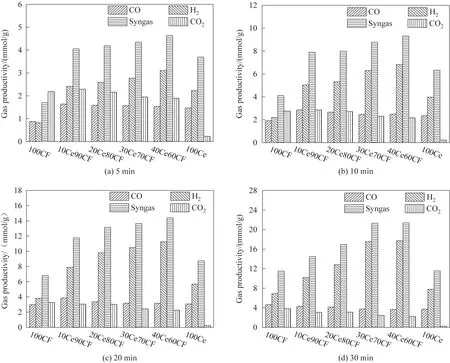
图8 CuFe2O4-CeO2与甲烷部分氧化不同阶段气体产量Fig.8 Gas yields of chemical looping partial oxidation of methane with CuFe2O4-CeO2 at different periods
图9 为不同CeO2掺杂量的氧载体与CH4部分氧化时反应性能评估。由图9(a)可知,CH4部分氧化反应最开始时转化率较大,随即以较快速率下降至最小值,又随着反应进行转化率逐渐上升。初始CH4转化率的快速下降主要归因于反应器中惰性气体对引入反应器中的CH4浓度的快速稀释;同时,氧载体表面氧浓度较高,对CH4转化率有较高的氧化活性。而当表面氧完全消耗时,氧载体内部晶格氧不断向表面迁移并参与反应,同时氧载体中出现被还原的金属单质Cu 的催化作用,CH4转化率逐渐上升并趋于稳定。当氧载体中晶格氧完全消耗时,CH4不断裂解产生积炭,沉积在氧载体表面导致氧载体反应活性衰减、CH4转化率下降。而随着CeO2掺杂含量增加,CH4部分氧化时从与纯CuFe2O4的最高转化率50%提高到30Ce70CF 时的80%。同时,CeO2加入促进了氧载体氧传递速率及反应活性的增加,使得CH4部分氧化产生合成气的时间提前,而且部分氧化阶段持续时间显著延长,从10Ce90CF 时的15 min 延迟到40Ce60CF 时的20 min,说明改性氧载体中CeO2及CuFe2O4之间的协同作用能有效提高CH4部分氧化阶段的转化率并促进了合成气形成时间的延长。

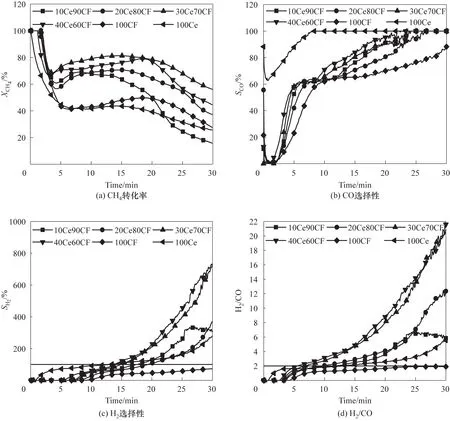
图9 CuFe2O4-CeO2与甲烷在900℃下恒温反应性能Fig.9 Reaction performance of methane with CuFe2O4-CeO2 oxygen carriers at 900℃
2.3 CO2恒温还原反应


图10 不同氧载体在900℃下与CO2还原性能Fig.10 Reduction performance of reduced CuFe2O4-CeO2 oxygen carriers with CO2 at 900℃
2.4 氧载体循环稳定性性能评价
图11 为30Ce70CF 氧载体5 次氧化还原循环实验,CH4部分氧化时间为15 min,CO2还原时间为20 min。图11(a)为氧载体循环反应中合成气产量,CH4部分氧化阶段合成气产量与CO2还原阶段CO 产量在前三次反应中逐渐增加,并在后续循环中保持稳定,表明氧载体在多次循环反应中具有较好的反应稳定性。图11(b)为循环反应时CH4部分氧化阶段的转化率、CO 选择性与H2/CO 比例。由图11(b)可见,第一次循环反应时,以30Ce70CF 为氧载体,CO与H2含量及合成气体产量较低,CH4转化率达到约86%,CO 选择性稳定在75%,而合成气H2/CO 比值为2.5,都相应地高于不掺杂CeO2的纯CuFe2O4的值。而在CO2还原阶段,由于前一阶段还原氧载体上CH4裂解沉积炭在CO2气氛下的气化,CO 产量大于10%,而且反应中CO 产量保持稳定,说明氧载体在连续循环中具有较高反应活性,而且随着循环反应次数增加,CeO2与CuFe2O4相互作用逐步增强,更紧密的接触降低了晶格氧传递阻力,增加了氧载体供氧能力,合成气产量增加并保持稳定。

图11 氧载体30Ce70CF循环反应Fig.11 Cyclic reactions of 30Ce70CF oxygen carrier
图12 为30Ce70CF 不同反应阶段的XRD 谱图。理论上,CuO、Fe2O3、CeO2可以被CH4还原为Cu、Fe和Ce2O3[30,39],而在实际反应过程中,与新鲜样品不同,经过第5 次CH4还原反应后,样品中仍有较强的CeO2特征峰,同时还检测到Ce2O3,而CuFe2O4尖晶石相的特征峰完全消失,检测到大量Cu 与Fe 单质的特征峰,这一结果表明,在CH4部分氧化阶段,CuFe2O4与CH4反应消耗了晶格氧,有部分Ce4+转化为Ce3+,同时还原单质Cu 的存在促进了Fe3+的深度还原形成了单质Fe,最后三者之间可能形成具有大量氧空位的Ce-Fe-Cu 固溶体,从而进一步增强了晶格氧传递能力与迁移速率,促进了30Ce70CF 反应活性的增强,因此与CH4部分氧化时反应时间明显缩短。

图12 30Ce70CF氧载体不同循环反应阶段XRD谱图Fig.12 XRD patterns of 30Ce70CF oxygen carrier at the different cyclic reaction stages
进一步,在与CO2部分氧化过程中,CeO2并没有大量转化,仍然保持原有晶相,并进一步与CuFe2O4还原形成的Fe离子结合生成钙钛矿结构CeFeO3,这种构型由于晶格重排中的电荷补偿和氧缺陷的形成,使得CeFeO3固溶体有助于提高氧的迁移率和产生氧空位[40-42],提高CuFe2O4的反应性,使其保持较高的CO选择性。同时,在CO2部分氧化中,由于CO2的弱氧化性,被还原的Cu 不能从CO2中获得晶格氧,Fe 不能被氧化为最高价态,最终产物中检测到Cu 单质和不高于Fe3O4的低价态Fe 氧化物特征峰,而还原阶段CeO2还原形成的Ce2O3也没有得以氧化。最后,经空气进一步氧化, 30Ce70CF 氧载体XRD 谱图与新鲜样品保持一致,说明空气氧化彻底恢复30Ce70CF 内部晶格氧,确保30Ce70CF 充分氧化再生。
最后,为了考察氧载体在反应中表面元素价态变化,以30Ce70CF 样品为例,对O 1s、Fe 2p、Ce 3d进行XPS 分析,XPS 谱图如图13 所示。在图13(a) O 1s 的XPS 谱图中,529 eV 附近的OI为晶格氧,531、533 eV 附近的OII、OⅢ分别为化学吸附氧和物理吸附氧,样品中较高的OI说明制备的样品晶格氧含量较高,在CH4部分氧化阶段因为CH4与晶格氧发生反应消耗导致OI峰值降低,在CO2还原阶段OI得以部分恢复,说明经过第5 次CO2部分氧化后,深度还原氧载体仍能获得较多晶格氧并保持一定的稳定性,但鉴于CO2弱氧化性,仍略低于新鲜样品,这与图12 XRD 对应的分析结果是一致的。在图13(b)中,Ce 3d的光谱可分为八个峰,分别记为V、V′、V″、V‴、U、U′、U″和U‴,其中V′和U′对应Ce3+的XPS峰,其余为Ce4+的XPS 峰。在CH4部分氧化阶段,V′和U′显示出比其他阶段更高的峰强,这说明CeO2向CuFe2O4提供了部分晶格氧参与反应,而在CO2还原阶段,V′和U′恢复到接近原始状态,说明部分消耗的晶格氧得以补充。而由图13(c) Fe 2p 谱图,在709 eV 附近的为Fe2+,理论上氧载体中全部为Fe3+,可能原因为处于表面的Fe 原子由于周围化学环境的变化,导致出现电子得失,部分Fe3+被还原为Fe2+。在CH4部分氧化阶段Fe3+更多被还原为Fe2+,甚至Fe单质;而在CO2还原阶段,Fe 单质消失,但仍有Fe2+存在。结合XRD 分析,CO2氧化并不能将失去的晶格氧完全恢复,因此空气氧化对于充分恢复氧载体的氧容量是非常必要的。

图13 氧载体30Ce70CF在CH4还原和CO2氧化过程中XPS谱图Fig.13 XPS maps of 30Ce70CF oxygen carrier in the CH4 reduction and CO2 catalytic oxidation stages
3 结 论
本文采用溶胶-凝胶燃烧合成法制备了掺杂不同含量CeO2的CuFe2O4氧载体,并用于CH4化学链部分氧化耦合CO2催化还原及空气氧化再生实验,通过XRD、BET、H2-TPR、SEM 等表征技术对新制备及不同反应阶段的氧载体的物理化学特性加以系统研究,发现CeO2的掺杂并没有改变CuFe2O4晶相,且掺杂后氧载体比表面积与孔体积均有所增大,并且随着掺杂量增加氧载体表面孔分布更加均匀,有助于更多活性位点的暴露,由此氧载体对CH4反应活性与释氧性能的强化大有裨益。
进一步,在固定床反应器中依序对不同掺杂量的CeO2强化的CuFe2O4混合氧载体在CH4部分氧化、CO2催化转化、空气氧化再生及多次循环反应性能测试表明,CeO2掺杂强化CuFe2O4氧载体产生大量氧空位,促进了氧载体释氧速率的快速提升,使得CH4转化率随CeO2掺杂量持续增加,但当CeO2掺杂过量后,CeO2晶粒团聚程度增加晶粒增大,与CuFe2O4不能充分接触,导致氧传递能力降低,反应活性下降,因此掺杂30%的CeO2为佳。
30Ce70CF 在连续5 次氧化还原循环后,CH4转化率稳定在86%,CO 选择性稳定在75%,而合成气H2/CO 比例约2.5,部分氧化阶段产生的积炭会被CO2气化,暴露更多活性位点促进了CO2充分催化还原,进一步提高CO 产量。最后,CO2部分氧化氧载体在空气中可以完全氧化再生。综上研究有助于CO2与CH4两种温室气体的资源化高值化利用。
